PROTEOME ANALYSIS
The term "Protein sequence analysis" in biology implies subjecting a peptide sequence to sequence alignment, sequence databases, repeated sequence searches, or other bioinformatics methods on a computer. Sequence analysis in molecular biology and bioinformatics is an automated, computer-based examination of characteristic fragments. It basically includes five biologically relevant topics, the comparison of sequences in order to find similar sequences (sequence alignment), prediction of protein structures, and comparison of homologous sequences to construct a molecular phylogeny.
The extraction, sorting and analyzing of sequence information about proteins is a major part of Bioinformatics. Sequence analysis is a process of trying to find something about a amino acid sequence, employing in-silico biology techniques. Due to rapid progress in algorithm development in computational biology, a large number of softwares are freely available that will compare unknown sequence to all of the sequence is available in the public domain. Currently, many of the international scientific journals require any newly discovered sequence to be submitted in a publicly available database before the discovery can be published. These new submitted sequences are then checked, annotated, cross-referenced and published. Each record is then curated and maintained in one of the many different databases available over the internet. The initial analysis carried out on the protein sequences are: composition analysis, molecular weight search, isoelectric point calculation, peptide mapping, hydrophobicity and hydrophilicity of the sequence, secondary structure prediction, fold prediction, transmembrance region prediction, coil structure prediction, signal peptide, motif and tertiary structure prediction etc. Once a new sequence is obtained from the database resources, then the following stepwise sequence analysis can be carried out.
There are mainly five major types for sequence analysis namely:
Type 1: Sequence retrieval and Preparation
Type 2: Similarity Searches and Phylogenic analysis
Type 3: Structure Prediction
Type 4: Profile and Pattern construction and search
Type5: Protein Function Prediction
TYPE -1: SEQUENCE RETRIEVAL AND PREPARATION:
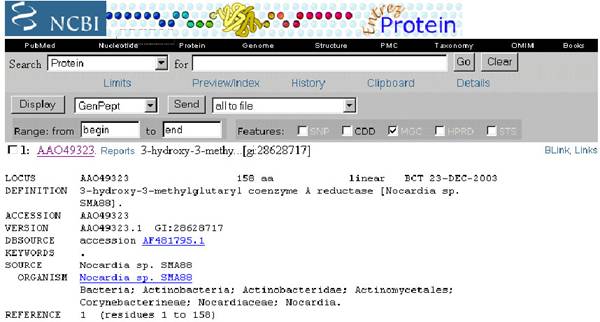
Protein sequence for the corresponding protein retrieved from the protein sequence databases like Genpept, Uniprot and PIR.
URL: http://www.ncbi,nlm.nih.gov/
For sequence analysis, different servers require different input sequence formats which can be obtained using sequence format converters. One of such server available in BCM is ReadSeq.
URL: http://searchlauncher.bcm.tmc.edu/seq-util/Options/readseq.html
The Homepage of the GenPept server is as follows:
The home page of the server is as follows:
TYPE-2 SIMILARITY SEARCHES AND PHYLOGENIC ANALYSIS:
Similarity searches were done through sequence alignment analysis. Sequence alignment is the process of lining up two or more sequences to achieve maximal levels of identity and conservation for the purpose of assessing the degree of similarity and the possibility of homology. Sequence similarity analysis is the single most powerful method for structural and functional inference available in databases. Sequence similarity analysis allows the inference of homology between proteins and homology can help one to infer whether the similarity in sequences would have similarity in function. Fundamentally, sequence-based alignment searches are string-matching procedures. A sequence of interest (query sequence) is compared with sequences (targets) in a databank-either pair-wise or with multiple target sequences, by searching for a series of individual characters. Two sequences are aligned by writing them across a page in two rows. Identical or similar characters are placed in the same column and non-identical characters can be placed opposite a gap in the other sequence. Gap is a space introduced into an alignment to compensate for insertions and deletions in one sequence relative to another. Due to this reciprocity between insertion and deletion, they are usually called indel for short. In optimal alignment, non-identical characters and gaps are placed to bring as many identical or similar characters as possible to vertical register.
The objective of sequence alignment analysis is to analyze sequence data to make reliable prediction on protein structure, functional and evolution vis a vis the three-dimensional structure. When character is shared between two species or populations, that character is said to be identical. The degree of which two species or populations share identities is indicated by similarity.
Mainly sequence alignment is studied under two headings namely Pair-wise sequence alignment and Multiple sequence alignment.
Pair-wise sequence alignment:
Pair-wise alignment is a fundamental process in sequence comparison analysis. Pair-wise alignment of two sequences is relatively straightforward computational problem. In a pair-wise comparison, if gaps or local alignments are not considered, the optimal alignment method can be tried and the number of computations required for two sequences is roughly proportional to the square of the average length, as is the case in dot plot comparison. The problem becomes complicated, and not feasible by optimal alignment method, when gaps and local alignment considered. A maximum match between two sequences is defined to be the largest number of amino acids from on protein that can be matched with those of another problem, while allowing for all possible deletions. A penalty is introduced to provide a barrier to arbitrary gap insertion. Pair-wise alignment achieved in two ways namely Local and Global alignment.
Local Alignment:
Local alignment is an alignment of some portion of two nucleic acid or protein sequences. Smith-Waterman algorithm is best alignment method for sequences for which no evolutionary relatedness is known. The program finds the region or regions of highest similarity between two sequences, thus generating one or more islands of matches or sub-alignments in the aligned sequences. Local alignments are more suitable and meaningful for aligning sequences that are similar along some of their lengths but dissimilar in others, sequences that differ in length or sequences that share conserved regions or domains.
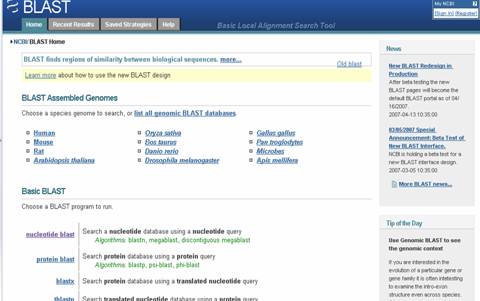
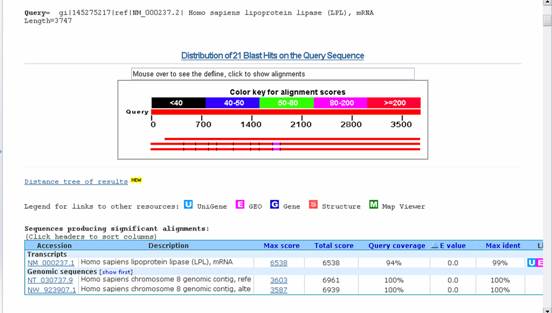
1. BLAST (Basic Local Alignment Search Tool) is one of the online tools available in NCBI. It is a popular user-friendly search tool for searching all the major sequence databases. It is a heuristic method to find the highest scoring locally optimal alignment between a query sequence and a database sequence. Blast programs are designed for fast database searching with minimal sacrifice of sensitivity to distant related sequences. It shows better results for protein sequences than nucleotide sequences. The default database is the non-redundant database, but the user still has the option to select one of their choices. The use of filters reduces problems of contamination with numerous artifacts in the databases.
URL: http://ncbi.nlm.nih.gov/blast/
Output:
Global Alignment:
Global alignment is an alignment of two nucleic acid or protein sequences over their entire length. The Needleman-Wunsch algorithm (GAP program) is one of the methods to carry out pair-wise alignment of sequences by comparing a pair of residues at a time. Comparisons are made from the smallest unit of significance, a pair of amino acids, one from each protein. All possible pairs are represented by a two-dimensional array and pathways through the array represent all possible comparisons. Statistical significance is determined by employing a scoring system; for a match=1 and mismatch=0 and penalty for a gap. Each cell in the matrix is examined, maximum score along any path leading to the cell is added to its present contents and the summation is constructed. In this way the maximum match pathway is constructed. The maximum match is the largest number that would result from summing the cell score values of every pathway, which is defined as the optimal alignment. Leaps to the non-adjacent diagonal cells in the matrix indicate the need for gap insertion, to bring the sequence into register. Complete diagonals of the array contain no gaps. Needleman algorithm tries to take all the characters of one sequence and align it with all the characters of a second sequence. This algorithm works well for sequences that show similarity across most of their lengths.
1. EMBOSS: Pair-wise alignment tool is utilized for global alignment.
URL: http://www.ebi.ac.uk/emboss/align/
Input:
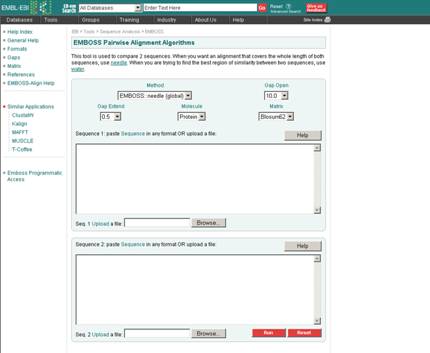
Output:

MULTIPLE SEQUENCE ALIGNMENT:
Multiple sequence alignment is an alignment of three or more sequences with gaps inserted in the sequences such that residues with common structural positions and/or ancestral residues are aligned in the same column. At the heart of the sequence analysis method is the multiple alignments. This is because of the quantity and diversity of information in databases. Pair-wise alignments are fundamental and useful, but there are some problems with them. For instance, when using one of other popular sequence searching programs like BLAST which perform pair-wise alignments to find similar sequences in a database, one very often obtains many sequences that are significantly similar to the query sequence. Comparing each and every sequence to every other may be possible when one has just a few sequences, but it quickly becomes impractical as the number sequences increases. But in multiple sequence alignment, all similar sequences can be compared in one single figure or table. The basic idea is that the sequences are aligned on top of each other, so that a coordinate system is set p, where each row is the sequences for one protein, and each column is the same position in each sequence. Each column corresponds to a specific residue in the prototypical protein. As with pair-wise alignment, there will be gaps in some sequence, most often was shown by dash ‘-‘or dot ‘.’ character. Note that to construct a multiple alignment; one may have to introduce gaps in sequences at positions where there were no gaps in the corresponding pair-wise alignment. This means that multiple alignments typically contain more gaps than any given pair of aligned sequences.
1. ClustalW:
ClustalW is one of the standard programs implementing one variant of the progressive method in wide use for multiple alignments. The W denotes a specific version that has been developed from the original clustal program. The basic steps of the algorithm implemented in clustalW are:
ClustalW is an example of an algorithm that has given up on trying to be perfect and instead uses an approximation strategy, combined with more or less intelligent tricks that guide the computation towards a successful result. This is called a heuristic algorithm.
One important point to keep in mind is that since clustalW is a heuristic algorithm, it cannot produce a solution that is guaranteed to be optima. But in practice, the result produces are good enough.
URL: http://www.ebi.ac.uk/clustalw/
Input:
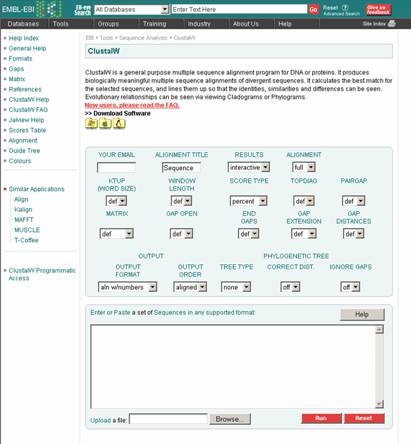
Output:

Phylogenetic Analysis:
Phylogenetic analysis of a family of related sequences is a determination of how the family might have been derived during evolution. Placing the sequences as outer branches on a tree depicts the evolutionary relationships among the sequences. The branching relationships on the inner part of the tree then reflect the degree to which different sequences are related. There are five different ways in which phylogenetic analysis carried out namely Dayhoff mutation data matrix method, Block model, Clustering algorithm, Distance method and Cladistic method. Phylogenetic trees are graphical representation of genetic relationships and the evolutionary history of taxa or sequences. The separated sequences are referred to as taxa, defined as phylogenetically distinct units on the tree.
The tree is composed of outer branches, representing the taxa and nodes and branches representing relationships among the taxa. Distance, maximum parsimony and maximum likelihood methods are generally used to find the evolutionary trees. Multiple sequence alignment plays crucial role in phylogenetic tree construction. The method of converting MSA to a phylogenetic tree is used to reduce the problem of o multiple alignment to an iterative process of pair-wise alignments. The process work as follows: compute all pair-wise distances between given sequences compute a tree by single linkage clustering by using methods like UPGMA or Nearest Neighbor and align the sequences in an orderly fashion.
There are various programs available for performing various phylogenetic operations. Different programs and program options are different for DNA and protein sequences.
1. Phylip (Phylogeny Inference Package) is a package of programs for inferring phylogenies. Methods supported in the package include parsimony, distance matrix, and likelihood methods, including bootstrapping and consensus trees. Data types that can be handled include molecular sequences, gene frequencies, restriction sites, distance matrices and 0/1 discrete characters.
URL: http://evolution.genetics.washington.edu/phylip.html
TYPE3 STRUCTURE PREDICTION:
Protein structure prediction from a sequence is one of the high focus problems for researchers. This is a very useful application of bioinformatics as the experimental techniques like X-ray crystallography are time consuming. The fundamental issue is how we can predict the 3-D shape of a protein from its amino acid sequence. This issue is solved in bioinformatics by following different algorithms and methods. Protein structure prediction was achieved in three different levels mainly Primary structure analysis, Secondary structure prediction and Tertiary structure prediction.
Primary structure analysis:
There are various tools for predicting the physical properties using the sequence information.
1. SAPS (Statistical Analysis of Protein Sequences):
Input:
SAPS is program that provides extensive statistical information for any given sequence. The output is organized in the following sections: file name, sequence printout, compositional analysis, charge distributional analysis, distribution of other amino acid types, repetitive structures, multiplets, periodicity analysis, and spacing analysis. The output is several pages long.
URL: http://www.isrec.isb-sib.ch/software/SAPS_form.html
2. ProtScale :
ProtScale allows you to compute and represent the profile produced by any amino acid scale on a selected protein. An amino acid scale is defined by a numerical value assigned to each type of amino acid.
The most frequently used scales are the hydrophobicity or hydrophilicity scales and the secondary structure conformational parameters scales, but many other scales exist which are based on different chemical and physical properties of the amino acids. This program provides 55 predefined scales entered from the literature.
URL: http://ca.expasy.org/protscale.pl
Input:
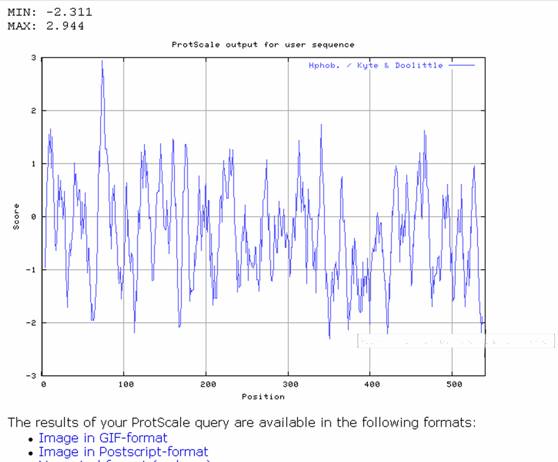
Output:
Secondary structure prediction:
There are several protein secondary structure prediction methods available and the most important methods are Chou-Fasman method, GOR methods, Nearest neighbour methods, Hidden Markov models, Neural networks and Multiple alignments based self-optimization method.
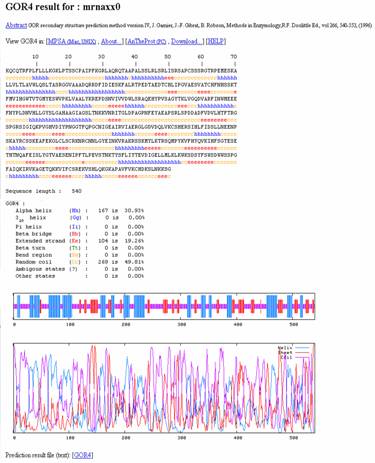
1. GOR (Garnier, Osguthorpe and Robson) method:
GOR is a method that assumes that amino acids up to 8 residues on each side influence the secondary structure of the central residue. This program is now in its fourth version. The accuracy of GOR when checked against a set of 267 proteins of known structure is 64%. This implies that 64% of the amino acids were correctly predicted as being helix, sheet or coil. The algorithm uses a sliding window of 17 amino acids. All possible pairs of amino acids in this window are checked for their information content as to predicting the structure of the central amino acid by comparing them to a set of 266 other proteins of known structure. The method works better for helix than for sheet, because sheet is dependent on longer-range interactions between non-adjacent sequence fragments.
URL: http://npsa-pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_gor4.html
Input:
Output:
2. COILS:
Coils is a program that compares a sequence to a database of known parallel two-stranded coiled coils and derives a similarity score. After comparing this score to the distribution of scores in globular and coiled-coil proteins, the program then calculates the probability that the sequence will adopt a coiled-coil conformation.
URL: http://www.ch.embnet.org/software/COILS_form.html
Input:
Output:
Tertiary Structure Prediction:
There are three fundamental approaches in using the sequence data for making protein structure prediction. Two approaches namely Homology modeling and Threading uses pattern recognition methods. The pattern recognition approach is used to detect similarity between sequences. This gives indications to infer related structures and functions. The other approach is to the sequence data without any template. This approach is called ab initio prediction. Ab initio approach is truly a prediction approach and is used to deduce structure and infer function directly from sequence.
Homology Modeling:
The prediction of protein three-dimensional (3D) structure from amino acid sequence is most successful when the structures of one or more homologues are known. A similar structure to the native structure can be predicted if sequence similarity is about or above 35%. Structural information can then be extrapolated to the new sequence and a 3D model may be derived, well before X-ray crystallography or NMR determines the structure of the new protein. This approach is most appropriately known as comparative modeling, but it also referred to as homology modeling or knowledge based modeling.
1. SWISS-MODEL:
It is a fully automated protein structure homology-modeling server, accessible via the ExPASy web server, or from the program DeepView (Swiss Pdb-Viewer). It goes through the following 5 steps : search for suitable templates, check sequence identity with target, create ProMod jobs, Generate models with ProModI and Energy minimization with Gromos96.
URL: http://swissmodel.expasy.org//SWISS-MODEL.html
Input1:

Input2:
Result of SWISS MODEL provided only through mail.
Threading:
When the sequence of a query protein has no detectable similarity to other protein structures (<20%), other methods of 3-D protein structure prediction may be enlarged. One such method is sequence threading. It is otherwise known as remote homology modeling or Fold Recognition modeling. Threading involves placing or “threading” an amino acid sequence onto databases of different secondary and tertiary structures. In effect, the procedure is aimed at predicting how well a fold will fit a sequence rather than predicting how well a sequence will fold. A target sequence is “threaded” through a library of 3-D folds to try to find a match. There are two approaches- 2-D threading and 3-D threading. 2-D threading is a “prediction-based” method that uses secondary structure as the primary evaluation criterion. The 3-D threading uses distance-based or profile-based energy functions. The amino acid sequence of a query protein is examined for compatibility with the structure core of a known protein structure. The sequence is threaded into a database of protein cores to look for matches.
URL: http://www.sbg.bio.ic.ac.uk/~3dpssm/
Input:
Abinitio Approach:
In contrast to the above methods, the goal of ab initio prediction is to build a model for a given sequence without using a template. Ab initio prediction relies on the thermodynamic hypothesis of protein folding. The ab initio prediction methods are based on the premise that the native structure of a protein sequence corresponds to its global free energy minimum state. Accoringly, the methods are generally formulated as optimizations. Molecular mechanics and molecular dynamics are used extensively in this type of structure prediction.
HMMSTR /I-sites/Rosetta Prediction Server:
This server predicts the tertiary structure of proteins from the sequence. I-sites predicts local structure, expressed as backbone torsion angles, using a library of sequence-structure motifs. ROSETTA is a Monte Carlo Fragment Insertion protein folding program. HMMSTR is HMM-based tool for local and secondary structure prediction based on I-sites Library. This server provide structure only if no homology present in databases.
URL: http://www.bioinfo.rpi.edu/~bystrc/hmmstr/server.php
Input:
Quarternary structure Prediction:
Quarternary structure deals with the specific arrangement of subunits(polypeptides), with respect one another in the protein compelx. Oligomeric protein usually possess quarternary structure.
Quarternary Structure Predictor:
The term "mericity" is used here to refer to "the number of subunits in a multisubunit protein". Mericity is a quaternary property of proteins. This experimental server accepts a query protein sequence (in single letter amino acid code form) and returns a prediction of the class (homodimer or non-homodimer, i.e. mericity=2 or not 2), a rule number, and a rule confidence level. A particular sequence may be covered by more than one rule. The true error rate of this classifier is approximately 30 per cent as determined by a 10-fold cross-validation experiment. These rules are highly sensitive to certain patterns in sequences, therefore, fragments and artificial sequences may give misleading results. It predicts the ability of the polypeptide to form homo or hetero dimmers.
Input:

Structure Validation:
This step is an important one in connection with structure prediction because predicted structure should be validated for its acceptance. Structure validation provided by servers like WHAT IF and Vadar.
WHAT IF:
The program WHAT IF provides nearly 2000 options in fields as diverse as homology modelling, drug docking, electrostatics calculations, structure validation and visualisation. This set of servers gives everybody access to some of these options. It provides its validation in the form of text file which contain ok, warning and error information about the protein structure. At the end of the validation message score given to the predicted protein.
Input:
Output:

VADAR:
VADAR (Volume, Area, Dihedral Angle Reporter) is a compilation of more than 15 different algorighms and programs for analyzing and assessing pepetide and protein structures from their PDB coordinate data. The results have been validated through extensive comparison to published data and careful visual inspection. The VADAR web server supports the submission of either PDB formatted files or PDB accession numbers. VADAR produces extensive tables and high quality graphs for quantitatively and qualitatively assessing protein structures determined by X-ray crystallography, NMR spectroscopy, 3D-thrading or homology modeling.
URL: http://redpoll.pharmacy.ualberta.ca/vadar/
Input:

Output:

TYPE 4 -PROFILE AND PATTERN CONSTRUCTION AND SEARCH:
Profiles:
Profiles are mathematical representation of conserved regions that are built from multiple sequence alignment. Profiles encompass full domain alignments, by defining which residues are allowed at given positions in the sequence, which positions are highly conserved and which positions/regions tolerate insertions.
Profiles program has four components namely an assembly of a family of related sequences into a multiple sequence alignment, construction of a profile from alignment, comparison of the profile to a database and display of the best similarity found with search. Profiles help to find the similarities between these sequences and help in identification and analysis of distant related proteins.
A position specific scoring table (PSSM) is constructed on the lines of PAM or BLOSUM. Much of the profile based search programs are based on statistical method, called Hidden Markov models (HMMs). Hidden Markov model is a Markov chain, which offers a more systematic approach to estimating parameters for domain alignments, by employing position dependent scores to characterize and build a model for an entire family of sequences.
ProfileScan:
It uses a database of profiles to find structural and sequence motifs in protein sequences. ProfileScan finds structural and sequence motifs in protein sequences. These motifs are represented as profiles in a library. ProfileScan aligns each profile motif to the sequence and displays all alignments between the profile and sequence that have a normalized score above a set threshold.
URL: http://hits.isb-sib.ch/cgi-bin/PFSCAN
Input:
Patterns:
Patterns also represent the common characteristics of a protein family, but it does not contain any weighting information. Pattern recognition programs follow reverse process of sequence analysis. Rather than predict how a sequence will fold, they predict how well a fold will match a sequence. That is, matching of sequence with a given topology rather than search for a topology with a given sequence. Pattern recognition methods attempt to detect similarities between 3-D structures that are not accompanied by any significant sequence similarity. The general approach involves calculating of a table of propensities that gives the probability for each type of amino aid being found in a given environment. For a given structure each position can be assigned to one of the environments. Dynamic programming is then used to find the best match of the sequence to the pattern of environments found in a given fold.
PRATT:
Pratt is a tool that allows the user to search for patterns conserved in a set of protein sequences. The user can specify what kind of patterns should be searched for, and how many sequences should match a pattern to be reported. The patterns that can be found is a subset of the set of patterns that can be described using Prosite notation.
URL: http://expasy.org/tools/pratt/
Input:
Profile and Pattern search:
URL: http://motif.genome.jp/MOTIF2.html
In this site, motifs, Profiles and patterns are searched and profiles generated. This server not only finds out sequence motifs in your query sequence, but also provides functional and genomic information of the found motifs using DBGET and LinkDB as the hyperlinked annotations. The results will also be presented graphically, and especially, where available, 3D structures of the found motifs can be examined by RasMol program when the hits are found in PROSITE database. Given a profile which was generated from the multiple sequence alignment, or, retrieved from motif library such as PROSITE or Pfam, one can align a protein sequence with the profile. The procedure is similar to the one to search against the motif library database, however, one should provide a name of the file containing profile matrix instead of the database names.
This server also supports TRANSFAC database which collects eukaryotic cis-acting regulatory DNA elements and trans-acting factors. Given a profile, protein sequence databases on GenomeNet service are retrieved to find out the protein families that have the same motif.
The profile, either in PROSITE or Pfam format, could be calculated from the multiple sequence alignment or retrieved from motif library such as PROSITE or Pfam. The Pfsearch program is used to retrieve with PROSITE format profile and Hmmsearch is used for Pfam format one. Target sequence libraries are Swiss-Prot, PDBSTR, PIR, PRF and Genes. This allows one to search protein sequence libraries with given patterns. Target sequence libraries are Swiss-Prot, PDBSTR, PIR, PRF and Genes. Sequence pattern must be specified in the PROSITE pattern format only. Two types of profile data, either in PROSITE or Pfam format, are calculated from the multiple alignment sequences. using PFMake or HMMBuild respectively.
TYPE5 PROTEIN FUNCTION PREDICTION:
Protein sequence determines protein structure determines protein function. Therefore, for function prediction, initially structure predicted and then function predicted. Predicting protein function from sequence adds two additional problems in comparison to the unsolved task of structure prediction:
These levels are related in complex ways e.g. protein kinases can be related to different cellular functions and to a chemical function plus a complex control mechanism by interaction with other proteins. Protein function prediction efforts generally involve attempts to predict biochemical function, cellular role predictions and subcellular location predictions.
ProtFun Server:
It produces ab initio predictions of protein function from sequence. The method queries a large number of other feature predictin servers to obtain information on various post-translational and localizatinal aspects of the protein, which are integrated into final predictions of the cellular role, enzyme class and selected gene ontology categories of the submitted sequence. It is possible to inspect the individual feature predictins used and integrated by ProtFun.
URL: http://www.cbs.dtu.dk/services/ProtFun/
Input:
EXPASY TOOLS
|
Protein identification and characterization |
|
Identification and characterization with peptide mass fingerprinting data |
|
|
Identification and characterization with MS/MS data |
|
|
Identification with isoelectric point, molecular weight and/or amino acid composition |
|
|
Other prediction or characterization tools |
|
|
Other tools for 2-DE or MS data (vizualisation, analysis, etc.) |
Make2D-DB II - A package to build a web-based proteomics database |
List of gene identification software sites - http://www.cbi.pku.edu.cn/mirror/GenomeWeb/nuc-geneid.html |
|
|
|
|
Topology prediction |
|
|
|
|
Tertiary structure analysis |
|
|
Tertiary structure prediction |
|
Comparative modeling
Threading
Ab initio
|
|
Assessing tertiary structure prediction |
|
|
Molecular modeling and visualization tools |
|
|
Prediction of disordered regions |
|
|
|
Binary |
|
|
Multiple |
|
|
Alignment analysis |
|
|
|