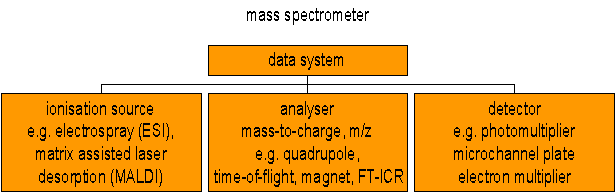
Simplified schematic of a mass spectrometer
An Introduction to Mass Spectrometry
Dr Alison E. Ashcroft,
Mass
Spectrometry Facility Manager,
Astbury Centre for
Structural Molecular Biology,
Astbury Building ,
The University of Leeds.
CONTENTS
Mass spectrometry is an analytical tool used for measuring the molecular mass of a sample.
For large samples such as biomolecules, molecular masses can be measured to within an accuracy of 0.01% of the total molecular mass of the sample i.e. within a 4 Daltons (Da) or atomic mass units (amu) error for a sample of 40,000 Da. This is sufficient to allow minor mass changes to be detected, e.g. the substitution of one amino acid for another, or a post-translational modification.
For small organic molecules the molecular mass can be measured to within an accuracy of 5 ppm or less, which is often sufficient to confirm the molecular formula of a compound, and is also a standard requirement for publication in a chemical journal.
Structural information can be generated using certain types of mass spectrometers, usually those with multiple analysers which are known as tandem mass spectrometers. This is achieved by fragmenting the sample inside the instrument and analysing the products generated. This procedure is useful for the structural elucidation of organic compounds and for peptide or oligonucleotide sequencing.
2. Where are mass spectrometers used?
Mass spectrometers are used in industry and academia for both routine and research purposes. The following list is just a brief summary of the major mass spectrometric applications:
3. How can mass spectrometry help biochemists?
4. How does a mass spectrometer work?
4.1 Introduction
Mass spectrometers can be divided into three fundamental parts, namely the ionisation source , the analyser , and the detector.
The sample has to be introduced into the ionisation source of the instrument. Once inside the ionisation source, the sample molecules are ionised, because ions are easier to manipulate than neutral molecules. These ions are extracted into the analyser region of the mass spectrometer where they are separated according to their mass (m) -to-charge (z) ratios (m/z) . The separated ions are detected and this signal sent to a data system where the m/z ratios are stored together with their relative abundance for presentation in the format of a m/z spectrum .
The analyser and detector of the mass spectrometer, and often the ionisation source too, are maintained under high vacuum to give the ions a reasonable chance of travelling from one end of the instrument to the other without any hindrance from air molecules. The entire operation of the mass spectrometer, and often the sample introduction process also, is under complete data system control on modern mass spectrometers.
Simplified schematic of a
mass spectrometer
4.2 Sample introduction
The method of sample
introduction to the ionisation source often depends on the ionisation method
being used, as well as the type and complexity of the sample.
The sample can be inserted directly into the ionisation source, or can undergo some type of chromatography en route to the ionisation source. This latter method of sample introduction usually involves the mass spectrometer being coupled directly to a high pressure liquid chromatography (HPLC), gas chromatography (GC) or capillary electrophoresis (CE) separation column, and hence the sample is separated into a series of components which then enter the mass spectrometer sequentially for individual analysis.
4.3 Methods of sample ionisation
Many ionisation methods are available and each has its own advantages and disadvantages ("Ionization Methods in Organic Mass Spectrometry", Alison E. Ashcroft, The Royal Society of Chemistry, UK, 1997; and references cited therein).
The ionisation method to be used should depend on the type of sample under investigation and the mass spectrometer available.
Ionisation methods include the following:
Atmospheric Pressure
Chemical Ionisation (APCI)
Chemical Ionisation (CI)
Electron Impact (EI)
Electrospray Ionisation (ESI)
Fast Atom Bombardment
(FAB)
Field Desorption / Field Ionisation (FD/FI)
Matrix
Assisted Laser Desorption Ionisation (MALDI)
Thermospray Ionisation
(TSP)
The ionisation methods used for the majority of biochemical analyses are Electrospray Ionisation (ESI) and Matrix Assisted Laser Desorption Ionisation (MALDI) , and these are described in more detail in Sections 5 and 6 respectively.
With most ionisation methods there is the possibility of creating both positively and negatively charged sample ions, depending on the proton affinity of the sample. Before embarking on an analysis, the user must decide whether to detect the positively or negatively charged ions (see section 7).
4.4 Analysis and Separation of Sample Ions
The
main function of the mass analyser is to separate
, or resolve , the ions formed in the ionisation
source of the mass spectrometer according to their mass-to-charge (m/z)
ratios. There are a number of mass analysers currently available, the
better known of which include quadrupoles ,
time-of-flight (TOF) analysers, magnetic sectors
, and both Fourier transform and quadrupole
ion traps .
These mass analysers have different features, including the m/z range that can be covered, the mass accuracy, and the achievable resolution. The compatibility of different analysers with different ionisation methods varies. For example, all of the analysers listed above can be used in conjunction with electrospray ionisation, whereas MALDI is not usually coupled to a quadrupole analyser.
Tandem (MS-MS) mass spectrometers are instruments that have more than one analyser and so can be used for structural and sequencing studies. Two, three and four analysers have all been incorporated into commercially available tandem instruments, and the analysers do not necessarily have to be of the same type, in which case the instrument is a hybrid one. More popular tandem mass spectrometers include those of the quadrupole-quadrupole, magnetic sector-quadrupole , and more recently, the quadrupole-time-of-flight geometries.
4.5 Detection and recording of sample ions.
The
detector monitors the ion current, amplifies it and the signal
is then transmitted to the data system where it is recorded in the form of
mass spectra . The m/z values of the ions are
plotted against their intensities to show the number of
components in the sample, the molecular mass of each
component, and the relative abundance of the various
components in the sample.
The type of detector is supplied to suit the type of analyser; the more common ones are the photomultiplier , the electron multiplier and the micro-channel plate detectors.
5. Electrospray ionisation
5.1 Electrospray
ionisation
Electrospray Ionisation (ESI) is one of
the Atmospheric Pressure Ionisation (API)
techniques and is well-suited to the analysis of polar molecules
ranging from less than 100 Da to more than 1,000,000 Da in molecular mass.
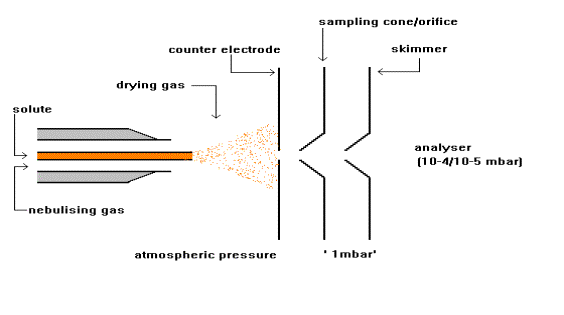
Standard electrospray
ionisation source (Platform II)
During standard electrospray ionisation (J. Fenn, J. Phys. Chem., 1984, 88, 4451), the sample is dissolved in a polar, volatile solvent and pumped through a narrow, stainless steel capillary (75 - 150 micrometers i.d.) at a flow rate of between 1 µL/min and 1 mL/min. A high voltage of 3 or 4 kV is applied to the tip of the capillary, which is situated within the ionisation source of the mass spectrometer, and as a consequence of this strong electric field, the sample emerging from the tip is dispersed into an aerosol of highly charged droplets, a process that is aided by a co-axially introduced nebulising gas flowing around the outside of the capillary. This gas, usually nitrogen, helps to direct the spray emerging from the capillary tip towards the mass spectrometer. The charged droplets diminish in size by solvent evaporation, assisted by a warm flow of nitrogen known as the drying gas which passes across the front of the ionisation source. Eventually charged sample ions, free from solvent, are released from the droplets, some of which pass through a sampling cone or orifice into an intermediate vacuum region, and from there through a small aperture into the analyser of the mass spectrometer, which is held under high vacuum. The lens voltages are optimised individually for each sample.
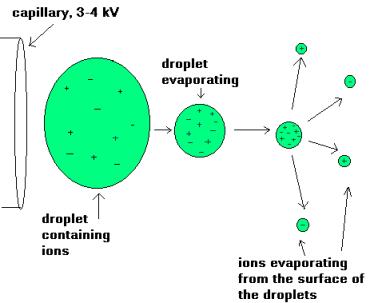
The electrospray
ionisation process
5.2 Nanospray
ionisation
Nanospray ionisation (M. Wilm, M. Mann,
Anal. Chem., 1996, 68, 1) is a low flow rate version of
electrospray ionisation. A small volume (1-4 microL) of the
sample dissolved in a suitable volatile solvent, at a concentration of ca.
1 - 10 pmol/microL, is transferred into a miniature
sample vial. A reasonably high voltage (ca.
700 - 2000 V) is applied to the specially manufactured gold-plated vial
resulting in sample ionisation and spraying. The flow rate of
solute and solvent using this procedure is very low, 30 - 1000
nL/min, and so not only is far less sample consumed than with the
standard electrospray ionisation technique, but also a small volume of sample
lasts for several minutes, thus enabling multiple experiments
to be performed. A common application of this technique is for a protein
digest mixture to be analysed to generate a list of molecular
masses for the components present, and then each component to be
analysed further by tandem mass spectrometric (MS-MS) amino acid
sequencing techniques (see Section 8).
ESI and nanospray ionisation are very sensitive analytical techniques but the sensitivity deteriorates with the presence of non-volatile buffers and other additives, which should be avoided as far as possible.
In positive ionisation mode, a trace of formic acid is often added to aid protonation of the sample molecules; in negative ionisation mode a trace of ammonia solution or a volatile amine is added to aid deprotonation of the sample molecules. Proteins and peptides are usually analysed under positive ionisation conditions and saccharides and oligonucleotides under negative ionisation conditions. In all cases, the m/z scale must be calibrated by analysing a standard sample of a similar type to the sample being analysed (e.g. a protein calibrant for a protein sample), and then applying a mass correction.
5.3 Data processing
ESI and
nanospray ionisation generate the same type of spectral data
for samples, and so the data processing procedures are identical.
In ESI, samples (M) with molecular masses up to ca. 1200 Da give rise to singly charged molecular-related ions, usually protonated molecular ions of the formula (M+H)+ in positive ionisation mode, and deprotonated molecular ions of the formula (M-H)- in negative ionisation mode.
An example of this type of sample analysis is shown in the m/z spectrum of the pentapeptide leucine enkephalin, YGGFL. The molecular formula for this compound is C28H37N5O7 and the calculated monoisotopic molecular weight is 555.2692 Da.
The m/z spectrum shows dominant ions at m/z 556.1, which are consistent with the expected protonated molecular ions, (M+H+). Protonated molecular ions are expected because the sample was analysed under positive ionisation conditions. These m/z ions are singly charged, and so the m/z value is consistent with the molecular mass, as the value of z (number of charges) equals 1. Hence the measured molecular weight is deduced to be 555.1 Da, in good agreement with the theoretical value.
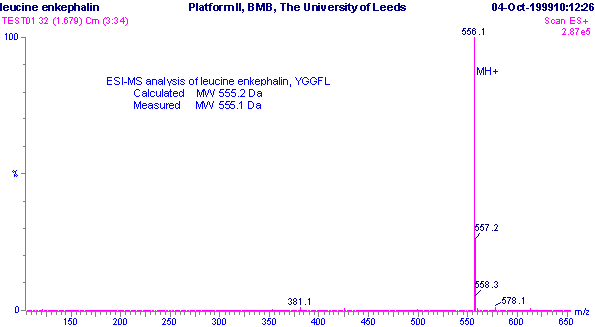
Positive ESI-MS m/z
spectrum of leucine enkaphalin, YGGFL.
The m/z spectrum also shows other ions of lower intensity (ca. 25 % of the m/z 556.1 ions) at m/z 557.2. These represent the molecule in which one 12C atom has been replaced by a 13C atom, because carbon has a naturally occurring isotope one atomic mass unit (Da) higher. The intensity of these isotopic ions relates to the relative abundance of the naturally occurring isotope multiplied by the total number of carbon atoms in the molecule. Additionally the fact that the 13C ions are one Da higher on the m/z scale than the 12C ions is an indication that z = 1, and hence the sample ions are singly charged. If the sample ions had been doubly charged, then the m/z values would only differ by 0.5 Da as z, the number of charges, would then be equal to 2.
The m/z spectrum also contains ions at m/z 578.1, some 23 Da higher than the expected molecular mass. These can be identified as the sodium adduct ions, (M+Na)+, and are quite common in electrospray ionisation. Instead of the sample molecules being ionised by the addition of a proton H+, some molecules have been ionised by the addition of a sodium cation Na+. Other common adduct ions include K+ (+39) and NH4+ (+18) in positive ionisation mode and Cl- (+35) in negative ionisation mode.
Electrospray ionisation is known as a "soft" ionisation method as the sample is ionised by the addition or removal of a proton, with very little extra energy remaining to cause fragmentation of the sample ions.
Samples (M) with molecular weights greater than ca. 1200 Da give rise to multiply charged molecular-related ions such as (M+nH)n+ in positive ionisation mode and (M-nH)n- in negative ionisation mode. Proteins have many suitable sites for protonation as all of the backbone amide nitrogen atoms could be protonated theoretically, as well as certain amino acid side chains such as lysine and arginine which contain primary amine functionalities.
An example of multiple charging, which is practically unique to electrospray ionisation, is presented in the positive ionisation m/z spectrum of the protein hen egg white lysozyme.
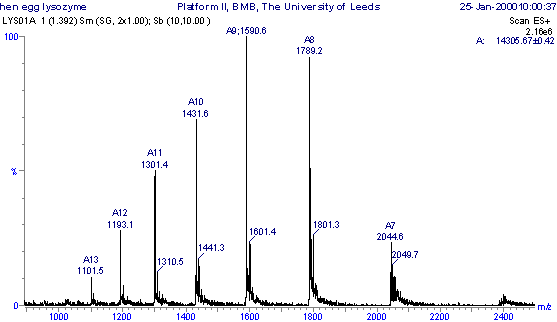
Positive ESI-MS m/z
spectrum of the protien hen egg white lysozyme.
The sample was analysed in a solution of 1:1 (v/v) acetonitrile : 0.1% aqueous formic acid and the m/z spectrum shows a Gaussian-type distribution of multiply charged ions ranging from m/z 1101.5 to 2044.6. Each peak represents the intact protein molecule carrying a different number of charges (protons). The peak width is greater than that of the singly charged ions seen in the leucine enkephalin spectrum, as the isotopes associated with these multiply charged ions are not clearly resolved as they were in the case of the singly charged ions. The individual peaks in the multiply charged series become closer together at lower m/z values and, because the molecular weight is the same for all of the peaks, those with more charges appear at lower m/z values than do those with fewer charges (M. Mann, C. K. Meng, J. B. Fenn, Anal. Chem., 1989, 61, 1702).
The m/z values can be expressed as follows:
m/z = (MW + nH+)/n
where m/z = the mass-to-charge ratio marked on the abscissa of the
spectrum;
MW = the molecular mass of the sample
n = the integer number of
charges on the ions
H = the mass of a proton = 1.008 Da.
If the number of charges on an ion is known, then it is simply a matter of reading the m/z value from the spectrum and solving the above equation to determine the molecular weight of the sample. Usually the number of charges is not known, but can be calculated if the assumption is made that any two adjacent members in the series of multiply charged ions differ by one charge.
For example, if the ions appearing at m/z 1431.6 in the lysozyme spectrum have "n" charges, then the ions at m/z 1301.4 will have "n+1" charges, and the above equation can be written again for these two ions:
1431.6 = (MW + nH+)/n and 1301.4 = [MW + (n+1)H+] /(n+1)
These simultaneous equations can be rearranged to exclude the MW term:
n(1431.6) - nH+ = (n+1)1301.4 - (n+1)H+
and so:
n(1431.6) = n(1301.4) +1301.4 - H+
therefore:
n(1431.6 - 1301.4) = 1301.4 - H+
and so:
n = (1301.4 -
H+) / (1431.6 - 1301.4)
hence the number of charges on the ions at m/z 1431.6 = 1300.4/130.2 = 10.
Putting the value of n back into the equation:
1431.6 = (MW + nH+) n
gives 1431.6 x 10 = MW + (10
x 1.008)
and so MW = 14,316 - 10.08
therefore MW = 14,305.9
Da
The observed molecular mass is in good agreement with the theoretical molecular mass of hen egg lysozyme (based on average atomic masses) of 14305.14 Da. The individual isotopes cannot be resolved when the ions have a large number of charges, and so for proteins the average mass is measured.
This may seem long-winded but fortunately the molecular mass of the sample can be calculated automatically, or at least semi-automatically, by the processing software associated with the mass spectrometer. This is of great help for multi-component mixture analysis where the m/z spectrum may well contain several overlapping series of multiply charged ions, with each component exhibiting completely different charge states.
Using electrospray or nanospray ionisation, a mass accuracy of within 0.01% of the molecular mass should be achievable, which in this case represents +/- 1.4 Da.
In order to clarify electrospray/nanospray data, molecular mass profiles can be generated from the m/z spectra of high molecular mass, multiply charged samples. To achieve this, all the components are transposed onto a true molecular mass (or zero charge state) profile from which molecular masses can be read directly without any amendments or calculations.
The m/z spectrum of lysozyme has been converted to a molecular mass profile using Maximum Entropy processing and the data are shown. The mass profile is dominated by a component of molecular mass 14,305.7 Da, with a series of minor peaks at higher mass, which is usually indicative of salt adducting e.g. Na (M+23), K (M+39), H2SO4 or H3PO4 (M+98). The molecular masses can be read easily and unambiguously, and a good idea of the purity of the protein is obtained on inspection of the molecular mass profile.
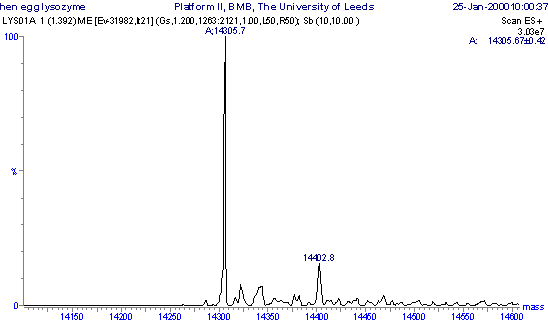
Molecular mass
profile of lysozyme obtained by maximum entropy processing of the m/z
spectrum
Proteins in their native state, or at least containing a significant amount of folding, tend to produce multiply charged ions covering a smaller range of charge states (say two or three). These charge states tend to have fewer charges than an unfolded protein would have, due to the inaccessibility of many of the protonation sites. In such cases, increasing the sampling cone voltage may provide sufficient energy for the protein to begin to unfold and create a wider charge state distribution centering on more highly charged ions in the lower m/z region of the spectrum.
The differences in m/z spectra due to the folded state of the protein are illustrated with the m/z spectra of the protein apo-pseudoazurin acquired under different solvent conditions.
Analysis of the protein in 1:1 acetonitrile : 0.1% aqueous formic acid at pH2 gave a Gaussian-type distribution with multiply charged states ranging from n = 9 at m/z 1487.8 to n = 19 at m/z 705.3, centering on n = 15 (lower trace). The molecular mass for this protein was 13,381 Da. Analysis of the protein in water gave fewer charge states, from n = 7 at m/z 1921.7 to n = 11 at m/z 1223.7, centering at n = 9 (upper trace). Not only has the charge state distribution changed, the molecular weight is now 13,444 Da which represents an increase of 63 Da and indicates that copper is remaining bound to the protein. Many types of protein complexes can be observed in this way, including protein-ligand, protein-peptide, protein-metal and protein-RNA macromolecules.
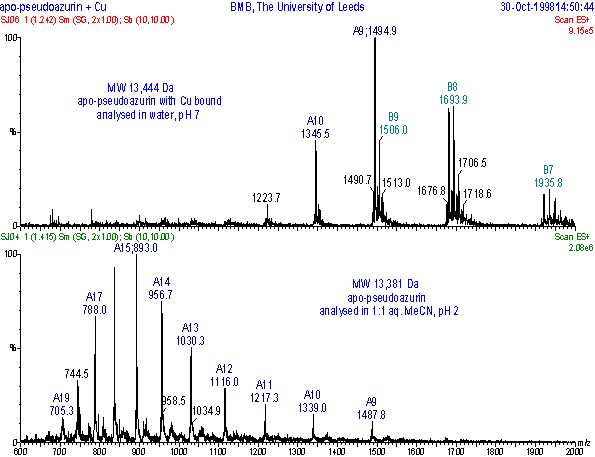
Positive ESI-MS m/z
spectra of the protein apo-pseudoazurin analysed in water at pH7 (upper trace)
and in 1:1 acetonitrile:0.1% aq. formic acid at pH2 (lower
trace).
6. Matrix assisted laser desorption ionisation
Matrix Assisted Laser Desorption Ionisation (MALDI) (F. Hillenkamp, M. Karas, R. C. Beavis, B. T. Chait, Anal. Chem., 1991, 63, 1193) deals well with thermolabile, non-volatile organic compounds especially those of high molecular mass and is used successfully in biochemical areas for the analysis of proteins, peptides, glycoproteins, oligosaccharides, and oligonucleotides. It is relatively straightforward to use and reasonably tolerant to buffers and other additives. The mass accuracy depends on the type and performance of the analyser of the mass spectrometer, but most modern instruments should be capable of measuring masses to within 0.01% of the molecular mass of the sample, at least up to ca. 40,000 Da.
MALDI is based on the bombardment of sample molecules with a laser light to bring about sample ionisation. The sample is pre-mixed with a highly absorbing matrix compound for the most consistent and reliable results, and a low concentration of sample to matrix works best. The matrix transforms the laser energy into excitation energy for the sample, which leads to sputtering of analyte and matrix ions from the surface of the mixture. In this way energy transfer is efficient and also the analyte molecules are spared excessive direct energy that may otherwise cause decomposition. Most commercially available MALDI mass spectrometers now have a pulsed nitrogen laser of wavelength 337 nm.
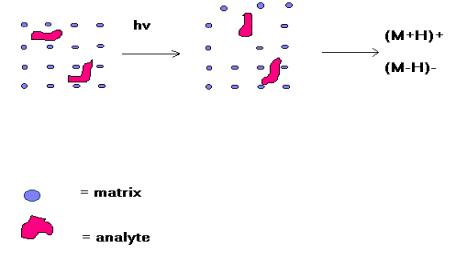
Matrix assisted laser
desorption ionisation (MALDI)
The sample to be analysed is dissolved in an appropriate volatile solvent, usually with a trace of trifluoroacetic acid if positive ionisation is being used, at a concentration of ca. 10 pmol/µL and an aliquot (1-2 µL) of this removed and mixed with an equal volume of a solution containing a vast excess of a matrix. A range of compounds is suitable for use as matrices: sinapinic acid is a common one for protein analysis while alpha-cyano-4-hydroxycinnamic acid is often used for peptide analysis. An aliquot (1-2 µL) of the final solution is applied to the sample target which is allowed to dry prior to insertion into the high vacuum of the mass spectrometer. The laser is fired, the energy arriving at the sample/matrix surface optimised, and data accumulated until a m/z spectrum of reasonable intensity has been amassed. The time-of-flight analyser separates ions according to their mass(m)-to-charge(z) (m/z) ratios by measuring the time it takes for ions to travel through a field free region known as the flight, or drift, tube. The heavier ions are slower than the lighter ones.
The m/z scale of the mass spectrometer is calibrated with a known sample that can either be analysed independently (external calibration) or pre-mixed with the sample and matrix (internal calibration).
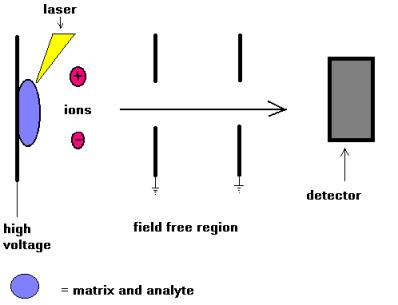
Simplified schematic
of MALDI-TOF mass spectrometry (linear mode)
MALDI is also a "soft" ionisation method and so results predominantly in the generation of singly charged molecular-related ions regardless of the molecular mass, hence the spectra are relatively easy to interpret. Fragmentation of the sample ions does not usually occur.
In positive ionisation mode the protonated molecular ions (M+H+) are usually the dominant species, although they can be accompanied by salt adducts, a trace of the doubly charged molecular ion at approximately half the m/z value, and/or a trace of a dimeric species at approximately twice the m/z value. Positive ionisation is used in general for protein and peptide analyses.
In negative ionisation mode the deprotonated molecular ions (M-H-) are usually the most abundant species, accompanied by some salt adducts and possibly traces of dimeric or doubly charged materials. Negative ionisation can be used for the analysis of oligonucleotides and oligosaccharides.
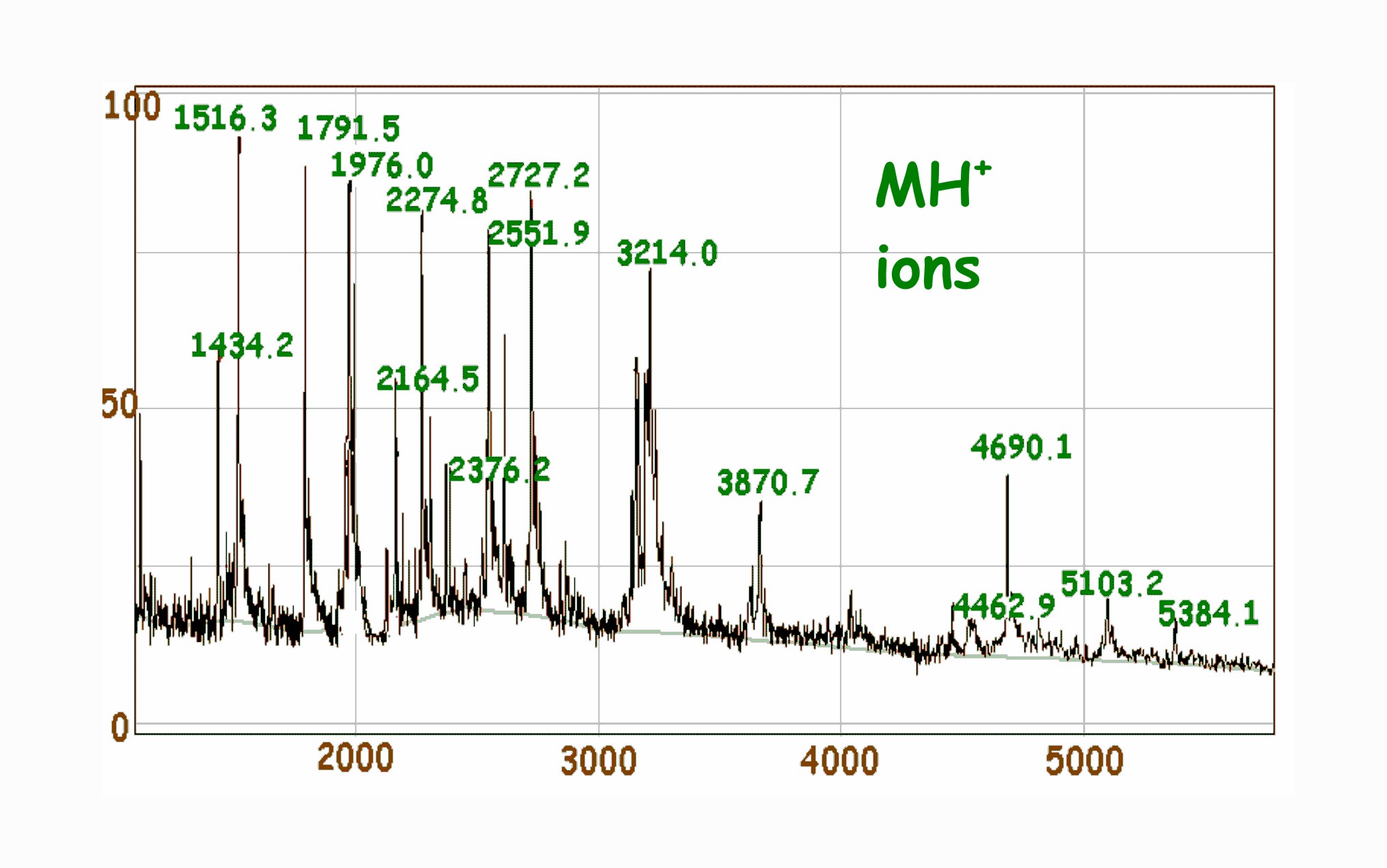
Positive
ionisation MALDI m/z spectrum of a peptide mixture using
alpha-cyano-4-hydroxycinnamic acid as matrix
7. Positive or negative ionisation?
If the sample has functional groups that readily accept a proton
(H+) then positive ion detection is used
e.g. amines
R-NH2 + H+ = R-NH3+ as in proteins
or peptides.
If the sample has functional groups that readily lose a proton
then negative ion detection is used
e.g. carboxylic acids
R-CO2H = R-CO2- and alcohols R-OH = R-O- as in
saccharides or oligonucleotides
8. Tandem mass spectrometry (MS-MS): Structural and sequence information from mass spectrometry.
8.1 Tandem mass
spectrometry
Tandem mass spectrometry (MS-MS)
is used to produce structural information about a compound by
fragmenting specific sample ions inside the mass spectrometer and identifying
the resulting fragment ions. This information can then be pieced together to
generate structural information regarding the intact molecule. Tandem mass
spectrometry also enables specific compounds to be detected in complex mixtures
on account of their specific and characteristic fragmentation patterns.
A tandem mass spectrometer is a mass spectrometer that has more than one analyser, in practice usually two. The two analysers are separated by a collision cell into which an inert gas (e.g. argon, xenon) is admitted to collide with the selected sample ions and bring about their fragmentation. The analysers can be of the same or of different types, the most common combinations being:
Fragmentation experiments can also be performed on certain single analyser mass spectrometers such as ion trap and time-of-flight instruments, the latter type using a post-source decay experiment to effect the fragmentation of sample ions.
8.2 Tandem mass spectrometry
analyses.
The basic modes of data acquisition for tandem mass
spectrometry experiments are as follows:
Product or daughter ion scanning:
the
first analyser is used to select user-specified sample ions arising from a
particular component; usually the molecular-related (i.e. (M+H)+ or
(M-H)-) ions. These chosen ions pass into the collision cell, are
bombarded by the gas molecules which cause fragment ions to be formed, and these
fragment ions are analysed i.e. separated according to their mass to charge
ratios, by the second analyser. All the fragment ions arise directly from the
precursor ions specified in the experiment, and thus produce a fingerprint
pattern specific to the compound under investigation.
This type of experiment is particularly useful for providing structural information concerning small organic molecules and for generating peptide sequence information.
Precursor or parent ion scanning:
the
first analyser allows the transmission of all sample ions, whilst the second
analyser is set to monitor specific fragment ions, which are generated by
bombardment of the sample ions with the collision gas in the collision cell.
This type of experiment is particularly useful for monitoring groups of
compounds contained within a mixture which fragment to produce common fragment
ions, e.g. glycosylated peptides in a tryptic digest mixture,
aliphatic hydrocarbons in an oil sample, or glucuronide
conjugates in urine.
Constant neutral loss scanning:
this
involves both analysers scanning, or collecting data, across the whole m/z
range, but the two are off-set so that the second analyser allows only those
ions which differ by a certain number of mass units (equivalent to a neutral
fragment) from the ions transmitted through the first analyser. e.g. This type
of experiment could be used to monitor all of the carboxylic acids in a mixture.
Carboxylic acids tend to fragment by losing a (neutral) molecule of carbon
dioxide, CO2, which is equivalent to a loss of 44 Da or atomic mass
units. All ions pass through the first analyser into the collision cell. The
ions detected from the collision cell are those from which 44 Da have been lost.
Selected/multiple reaction monitoring:
both of the analysers are static in this case as user-selected specific ions
are transmitted through the first analyser and user-selected specific fragments
arising from these ions are measured by the second analyser. The compound under
scrutiny must be known and have been well-characterised previously before this
type of experiment is undertaken. This methodology is used to confirm
unambiguously the presence of a compound in a matrix e.g. drug testing with
blood or urine samples. It is not only a highly specific method but also has
very high sensitivity.
8.3 Peptide Sequencing by Tandem Mass Spectrometry.
The most common usage of MS-MS in biochemical areas is the product or daughter ion scanning experiment which is particularly successful for peptide and nucleotide sequencing.
Peptide sequencing: H2N-CH(R')-CO-NH-CH(R")-CO2H
Peptides fragment in a reasonably well-documented manner (P. Roepstorrf, J. Fohlmann, Biomed. Mass Spectrom., 1984, 11, 601; R. S. Johnson, K. Biemann, Biomed. Environ. Mass Spectrom., 1989, 18, 945). The protonated molecules fragment along the peptide backbone and also show some side-chain fragmentation with certain instruments (Four-Sector Tandem Mass Spectrometry of Peptides, A. E. Ashcroft, P. J. Derrick in "Mass Spectrometry of Peptides" ed. D. M. Desiderio, CRC Press, Florida, 1990).
There are three different types of bonds that can fragment along the amino acid backbone: the NH-CH, CH-CO, and CO-NH bonds. Each bond breakage gives rise to two species, one neutral and the other one charged, and only the charged species is monitored by the mass spectrometer. The charge can stay on either of the two fragments depending on the chemistry and relative proton affinity of the two species. Hence there are six possible fragment ions for each amino acid residue and these are labelled as in the diagram, with the a, b, and c" ions having the charge retained on the N-terminal fragment, and the x, y", and z ions having the charge retained on the C-terminal fragment. The most common cleavage sites are at the CO-NH bonds which give rise to the b and/or the y" ions. The mass difference between two adjacent b ions, or y"; ions, is indicative of a particular amino acid residue (see Table of amino acid residues at the end of this document).
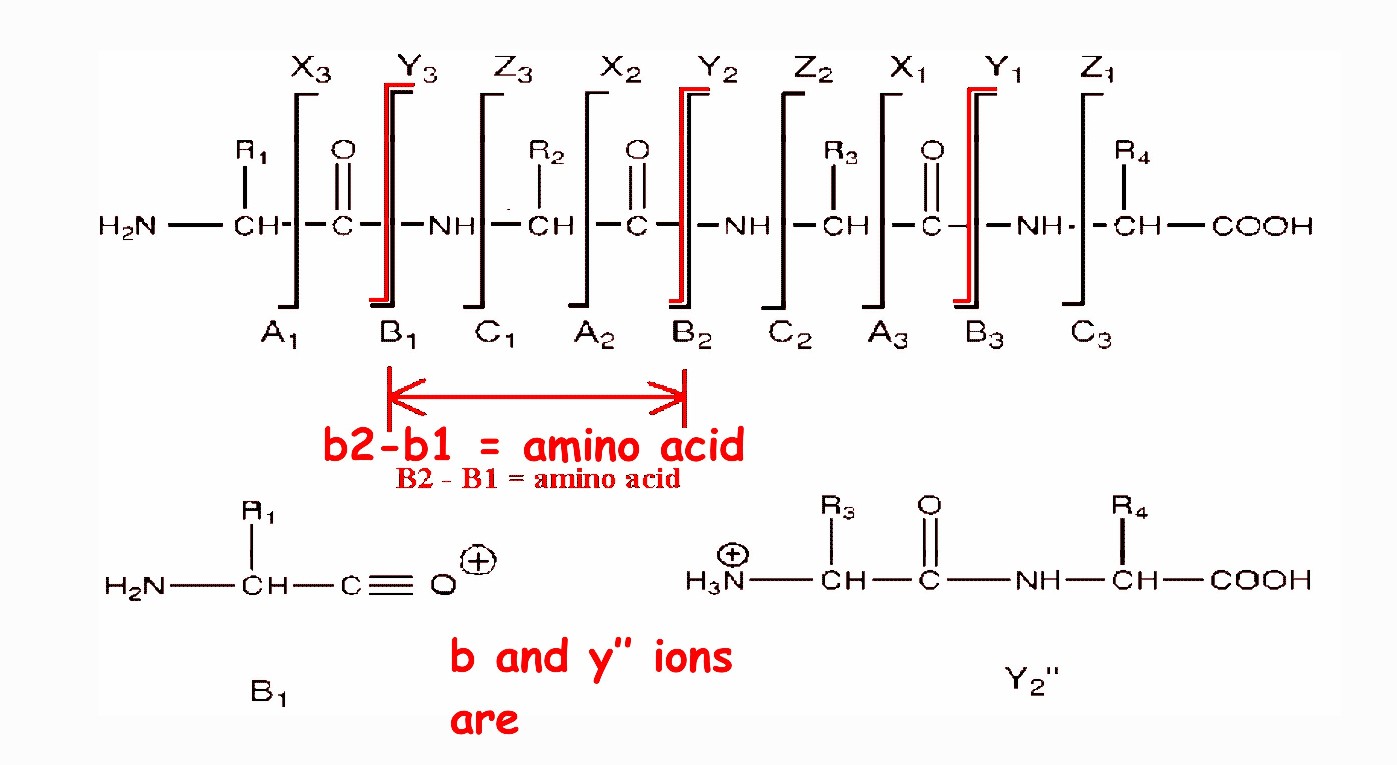
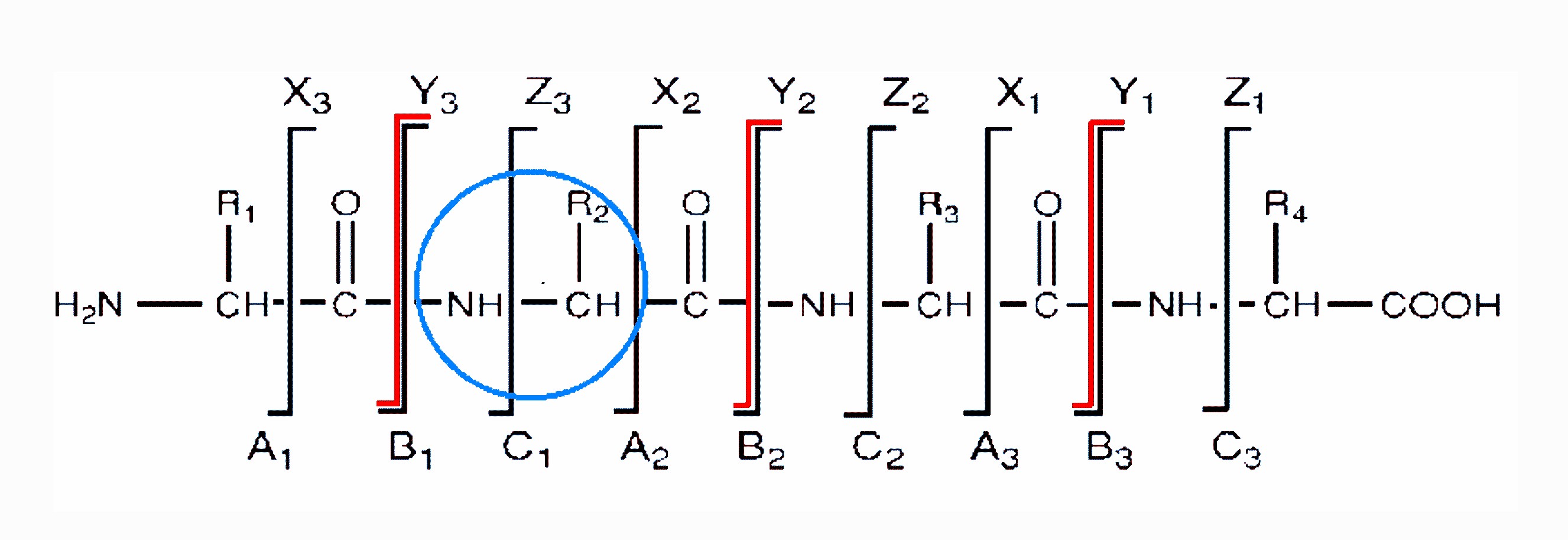
Peptide sequencing by tandem mass spectrometry -
backbone cleavages
The extent of side-chain fragmentation detected depends on the type of analysers used in the mass spectrometer. A magnetic sector - magnetic sector instrument will give rise to high energy collisions resulting in many different types of side-chain cleavages. Quadrupole - quadrupole and quadrupole - time-of-flight mass spectrometers generate low energy fragmentations with fewer types of side-chain fragmentations.
Immonium ions (labelled "i") appear in the very low m/z range of the MS-MS spectrum. Each amino acid residue leads to a diagnostic immonium ion, with the exception of the two pairs leucine (L) and iso-leucine (I), and lysine (K) and glutamine (Q), which produce immonium ions with the same m/z ratio, i.e. m/z 86 for I and L, m/z 101 for K and Q. The immonium ions are useful for detecting and confirming many of the amino acid residues in a peptide, although no information regarding the position of these amino acid residues in the peptide sequence can be ascertained from the immonium ions.
An example of an MS/MS daughter or product ion spectrum is illustrated below. The molecular mass of the peptide was measured using standard mass spectrometric techniques and found to be 680.4 Da, the dominant ions in the MS spectrum being the protonated molecular ions (M+H+) at m/z 681.4. These ions were selected for transmission through the first analyser, then fragmented in the collision cell and their fragments analysed by the second analyser to produce the following MS/MS spectrum. The sequence (amino acid backbone) ions have been identified, and in this example the peptide fragmented predominantly at the CO-NH bonds and gave both b and y" ions. (Often either the b series or the y" series predominates, sometimes to the exclusion of the other). The b series ions have been labelled with blue vertical lines and the y" series ions have been labelled with red vertical lines. The mass difference between adjacent members of a series can be calculated e.g. b3-b2 = 391.21 - 262.16 = 129.05 Da which is equivalent to a glutamine (E) amino acid residue; and similarly y4 - y3 = 567.37 - 420.27 = 147.10 Da which is equivalent to a phenylalanine (F) residue. In this way, using either the b series or the y" series, the amino acid sequence of the peptide can be determined and was found to be NFESGK (n.b. the y" series reads from right to left!). The immonium ions at m/z 102 merely confirm the presence of the glutamine (E) residue in the peptide.
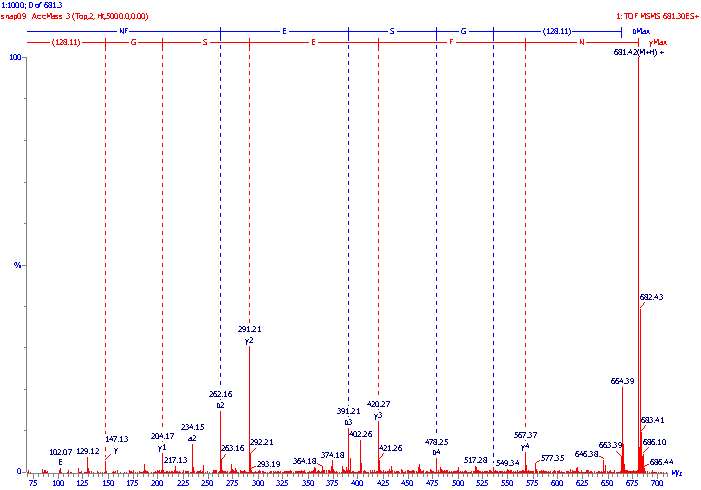
Peptide sequencing by
tandem mass spectrometry - an MS-MS daughter or product ion spectrum.
A protein identification study would proceed as follows:
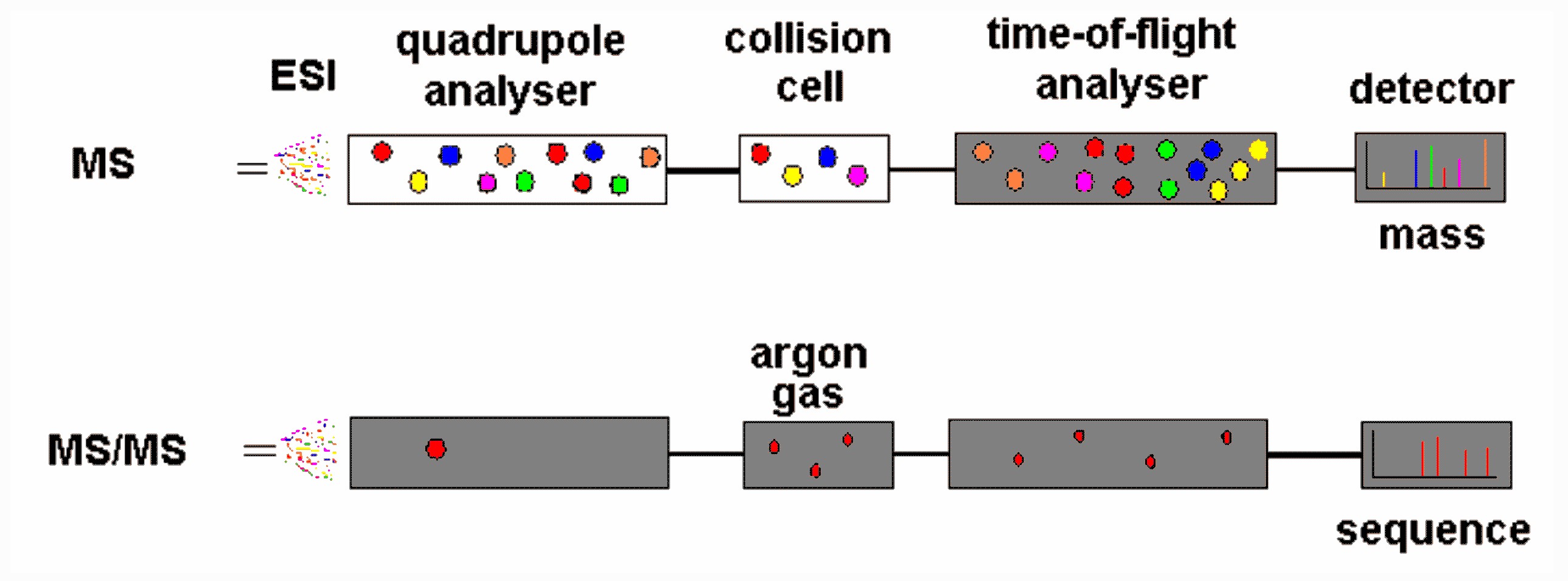
Q-TOF mass
spectrometer operating in MS (upper) and MS/MS mode (lower)
modes.
The amount of sequence information generated will vary from one peptide to another, Some peptide sequences will be confirmed totally, other may produce a partial sequence of, say, 4 or 5 amino acid residues. Often sequence "tag" of 4 or 5 residues is sufficient to search a protein database and confirm the identity of the protein.
Peptide sequencing in summary:
Peptides fragment along the amino acid backbone to give sequence information.
Peptides ca. 2500 Da or less produce the most useful data.
The amount of sequence information varies from one peptide to another. Some peptides can generate sufficient information for a full sequence to be determined; others may generate a partial sequence of 4 or 5 amino acids.
A protein digest can be analysed as an entire reaction mix, without any separation of the products, from which individual peptides are selected and analysed by the mass spectrometer to generate sequence information.
About 4 µL of solution is required for the analysis of the digest mixture, with a concentration based on the original protein of ca. 1-10 pmol/µL. MS/MS sequencing is a sensitive technique consuming little sample.
Sometimes the full protein sequence can be verified; some proteins generate sufficient information to cover only part of the sequence. 70 - 80% coverage is reasonable.
Often a sequence "tag" of 4/5 amino acids from a single proteolytic peptide is sufficient to identify the protein from a database.
The final point in this summary means that mass spectrometers have been found to be extremely useful for proteomic studies, as illustrated below.
The proteomics procedure usually involves excising individual spots from a 2-D gel and independently enzymatically digesting the protein(s) contained within each spot, before analysing the digest mixture by mass spectrometer in the manner outlined above. Electrospray ionisation or MALDI could be used at this step.
The initial MS spectrum determining the molecular masses of all of the components in the digest mixture can often provide sufficient information to search a database using just several of the molecular weights from this peptide map.
If the database search is not fruitful, either because the protein has not been catalogued, is previously uncharacterised, or the data are not accurate or comprehensive enough to distinguish between several entries in the database, then further information is required.
This can be achieved by sample clean-up and then MS/MS studies to determine the amino acid sequences of the individual proteolytic peptides contained in the digest mixture, with which further database searching can be carried out.
8.4 Oligonucleotide sequencing by Tandem Mass Spectrometry.
Oligonucleotide sequencing: P-S(B)-P-S(B)-P-S(B)
Oligonucleotide sequencing can also be achieved by tandem mass spectrometry although it is not so well documented. However fragmentation patterns have been established and reported (S. Pomerantz, J. A. Kowalak, J. A. McClosky, J. Amer. Soc. Mass Spectrom., 1993, 4, 204). The experimental principle is similar to that of peptide sequencing, in that individual species are mass measured in MS mode of instrument operation, and then their molecular-related ions selected by the first (quadrupole) analyser to be transmitted into the collision cell where they undergo fragmentation after bombardment with a collision gas. The fragments are analysed by the second (time-of-flight) analyser to produce an MS/MS product, or daughter, ion spectrum showing all the fragment ions that arise directly from the chosen parent or precursor ions.
Negative electrospray ionisation is often the preferred ionisation method. The optimisation of the fragmentation conditions varies from component to component and diligence must be taken to ensure the best conditions are employed.
Data processing and interpretation is again of paramount importance for accurate, reliable results and hence sequence information.
9. General reading
"Mass Spectrometry: A Foundation Course", K. Downard, Royal Society of Chemistry, UK, 2004.
"An Introduction to Biological Mass Spectrometry", C. Dass, Wiley, USA, 2002.
"The Expanding Role of Mass Spectrometry in Biotechnology", G. Siuzdak, MCC Press, San Diego, 2004.
"Ionization Methods in Organic Mass Spectrometry", A.E. Ashcroft, Analytical Monograph, Royal Society of Chemistry, UK, 1997.
http://www.astbury.leeds.ac.uk (A.E. Ashcroft's MS web pages and tutorial)
Table of amino acid residues .
| Symbol | Structure | Mass (Da) |
| Ala A | -NH.CH.(CH3).CO- | 71.0 |
| Arg R | -NH.CH.[(CH2)3.NH.C(NH).NH2].CO- | 156.1 |
| Asn N | -NH.CH.(CH2CONH2).CO- | 114.0 |
| Asp D | -NH.CH.(CH2COOH).CO- | 115.0 |
| Cys C | -NH.CH.(CH2SH).CO- | 103.0 |
| Gln Q | -NH.CH.(CH2CH2CONH2).CO- | 128.1 |
| Glu E | -NH.CH.(CH2CH2COOH).CO- | 129.0 |
| Gly G | -NH.CH2.CO- | 57.0 |
| His H | -NH.CH.(CH2C3H3N2).CO- | 137.1 |
| Ile I | -NH.CH.[CH.(CH3)CH2.CH3].CO- | 113.1 |
| Leu | -NH.CH.[CH2CH(CH3)2].CO- | 113.1 |
| Lys K | -NH.CH.[(CH2)4NH2].CO- | 128.1 |
| Met M | -NH.CH.[(CH2)2.SCH3].CO- | 131.0 |
| Phe F | -NH.CH.(CH2Ph).CO- | 147.1 |
| Pro P | -NH.(CH2)3.CH.CO- | 97.1 |
| Ser S | -NH.CH.(CH2OH).CO- | 87.0 |
| Thr T | -NH.CH.[CH(OH)CH3).CO- | 101.0 |
| Trp W | -NH.CH.[CH2.C8H6N].CO- | 186.1 |
| Tyr Y | -NH.CH.[(CH2).C6H4.OH].CO- | 163.1 |
| Val V | -NH.CH.[CH(CH3)2].CO- | 99.1 |