
Drugs
Antibacterial agents (antibiotics)
Antibacterial
antibiotics act at a variety of sites. However, in many cases, they act by
either making the plasma membrane of bacteria more permeable to essential ions
and other small molecules by ionophoric action or by inhibiting cell wall
synthesis. Those compounds that act on the plasma membrane also have the ability
to penetrate the cell wall structure. In both cases, the net result is a loss in
the integrity of the fungal cell envelope, which leads to irreversible cell
damage and death.
Ionophoric antibiotic action
Ionophores are
substances that can penetrate a cell membrane and increase its permeability
to ions. They may
be naturally occurring compounds such as the antibiotic gramicidin A produced by
Bacillus brevis
and valinomycin obtained from
Streptomyces fulvissimus, or synthetic compounds like the crown and
cryptate compounds (Fig.). However, ionophores transport ions in both directions
across a membrane. Consequently, they will only reduce the concentration of a
specific ion until its concentration is the same on both sides of a membrane.
However, a number of drugs are believed to owe their action to the ionophoric
transfer of essential substances out of the cell.
Examples of naturally occurring and synthetic ionophores
The general mode of action of ionophores in ion transport. (a)
A channel formed by two gramicidin A molecules, N-terminal to N-terminal. (b)
The sequence of events in the operation of a carrier ionophore such as
valinomycin
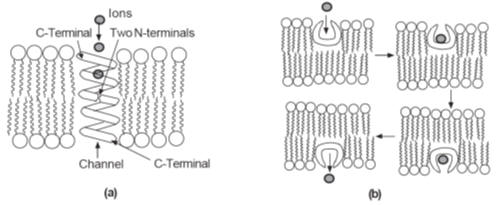
Ionophores
operate in two ways:
1. They form
channels across the membrane through which ions can diffuse down a concentration
gradient (Fig.a).
2. They act as
carriers that pick up the ion on one side of the membrane, transport it across,
and release it into the fluid on the other side of the membrane (Fig. b).
The structure of
each channel in a
Channel ionophore
is characteristic of the channel-former. For example, gramicidin forms a channel
(tube) composed of two molecules whose N-terminals meet in the middle of the
membrane.
Each gramicidine
molecule is in the form of a left-handed helix, which results in the polar
groups lining the interior of the channel. This facilitates the transfer of
polar ions through the channel. A single gramicidin channel can allow the
transport of up to 107 K+
ions per second.
Carrier
ionophores
are specific for particular ions. For example,
valinomycin will transport K+
ions but not Na2+
or Li+
ions. It is believed to form an octahedral complex with six carbonyl group
oxygen atoms acting as ligands. The resulting chelation complex has a
hydrophobic exterior that allows the complex to diffuse through the membrane.
However, the rigid nature of the molecule coupled with its size makes the
binding site of valinomycin too large to form octahedral complexes with the
smaller Na2+and
Li+
ions. Consequently, it
is more energetically favourable for Na2+
and Li+
ions to remain in solution as their hydrated ions. Ionophores are mainly active
against Gram-positive bacteria. However, until now, most of the compounds
examined do not significantly differentiate between bacterial and mammalian
membranes and so are of little clinical use. However, they are of considerable
use as research tools in the investigation of drug action.
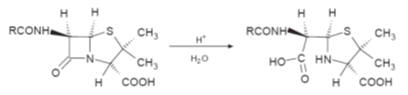
Cell wall synthesis inhibition
The cell walls of
all bacteria are being continuously replaced because they are continuously being
broken down by enzymes in the extracellular fluid. Antibacterial agents can
inhibit this replacement biosynthesis of the cell wall at any stage in its
formation. Investigations using
Staphylococcus aureus
have yielded a great deal of detail about the biosynthesis of its cell wall but
there are still areas of the biosynthesis that have not yet been fully
elucidated. Experimental investigations of the cell wall synthesis of other
bacterial species suggest that similar routes are followed. A detailed knowledge
of the route followed and the enzymes involved is an extremely useful
prerequisite in the quest for new drug substances.
It is convenient
to introduce the subject of antibacterial action due to inhibition of cell wall
synthesis by dividing the synthesis into three stages:
1. The formation
of precursor starting materials.
2. The formation
of the peptidoglycan chains.
3. The
cross-linking of the peptidoglycan chains.
However, it
should be realised that not only can an antibiotic inhibit cell wall formation
but it may also have other areas of action such as the plasma membrane of a
bacterium. Furthermore, it is emphasised that the biochemical pathways discussed
are a simplification based on experimental evidence. However, it is likely that
the drugs act in the same manner
on other
susceptible bacteria.
Drugs that inhibit the formation of the starting compounds
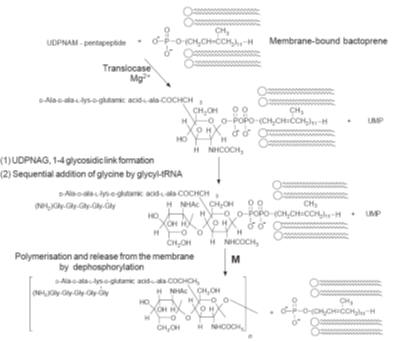
A convenient starting point for cell wall synthesis is
N-acetylglucosamine-1-phosphate
(NAG-1-P), which is found in all life forms. This compound is believed to react
with uridine triphosphate (UTP) to form uridine diphospho-N-acetylglucosamine
(UDPNAG), one of the precursors of the peptidoglycan chain (Fig.). Some of the
UDPNAG is further converted by a series of steps into the uridine diphospho-N
acetylmuramic acid pentapeptide derivative (UDPNAM-pentapeptide),
the second precursor of the peptidoglycan polymer chain. Drug action can inhibit
any of the steps in the formation of both UDPNAG and UDPNAM-pentapeptide.
However, inhibition of the synthesis of the latter is likely to be potentially
more rewarding since its formation requires a larger number of steps, which
gives a wider scope for intervention. Drugs in clinical use that act by
inhibiting different processes in this stage of cell wall synthesis are
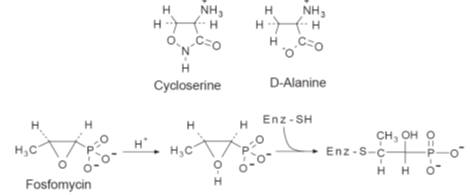
cycloserine and fosfomycin (Fig.).
Cycloserine and fosfomucin
An outline of the biosynthesis of the precursors of the peptidoglycan chain of
the cell wall of
Staphylococcus aureus

Cycloserine is a
broad spectrum antibiotic produced by
Streptomyces orchidaceus. The drug
is used mainly as
a second-line antitubercular agent. It enters the bacteria by active transport
systems, which results in a high concentration in the bacterial cell, a primary
requirement for activity. D-Cycloserine inhibits both alanine racemase and
D-alanyl-D-alanine synthetase, which blocks the conversion of the tripeptide to
the pentapeptide at two places (A in Fig.). The affinity of the enzymes in
Staphylococcus aureus
for the drug has been found to be 100 times higher than its affinity for its
natural substrate D-alanine. This affinity is believed to depend on the
isoxazole ring, whose shape corresponds to one of the conformations of
D-alanine. It is believed that the rigid structure of the isoxazole ring gives
the drug a better chance of binding to the active sites of the enzymes than the
more flexible structure of D-alanine.
An outline of the formation of the peptidoglycan chains of
Staphylococcus aureus
from UDPNAM-pentapeptide and
UDPNAG. UMP is uridine monophosphate
Fosfomycin,
produced by a number of Streptomyces species, is active against both
Gram-positive and Gram-negative bacteria. However, it is used mainly to treat
Gram-positive infections. The drug acts by inhibiting the enol-pyruvate
transferase (B in Fig.) that catalyses the incorporation of phosphoenolpyruvic
acid (PEP) into the UDPNAG molecule. However, the drug does not inhibit other
enol-pyruvate transferases used to incorporate PEP in a number of other
biosynthetic reactions. Consequently, it appears that the activity of the drug
is due to it forming an inactive product with the enzyme. It has been suggested
that this product is formed by the acid-catalysed nucleophilic substitution of
the oxiran ring by the sulphydryl groups of the cysteine residues in the active
site of the enzyme.
Drugs that inhibit the synthesis of the peptidoglycan chain
The sequence of reactions starting from UDPNAM-pentapeptide and UDPNAG to form
the peptidoglycan chain (Fig.) is not completely known although the main stages
have been identified. However, it is known that the reactions are catalysed by
membrane-bound enzymes. A number of antibiotics, such as bacitracin, are
believed to inhibit some of the stages of the biosynthesis of the peptidoglycan
chains.
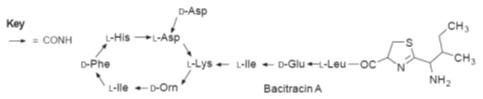
Bacitracin is a
mixture of similar peptides produced from
Bacillus subtilis.
The main component of this mixture is bacitracin A, which is active against
Gram-positive bacteria. However, its high degree of neuro- and nephrotoxicity
means that the drug is seldom used, and then somewhat cautiously. Its main site
of action appears to be inhibition of the dephosphorylation of membrane-bound
phospholipid carrier bactoprene (step M in Fig.). Its action is enhanced by the
presence of zinc ions.
Drugs that inhibit the cross-linking of the peptidoglycan chains
The final step in the formation of the cell wall is the completion of the
cross-links. This converts the water-soluble and therefore mobile peptidoglycans
into the insoluble stationary cell wall. Investigations using
Staphylcoccus aureus
indicated that the cross-linking is brought about by a multistep
displacement of the terminal alanine of the peptide attached to one
peptidoglycan chain and its replacement by the terminal glycine of the peptide
attached to a second peptidoglycan chain (Fig.). This reaction is catalysed by
transpeptidases.
An schematic outline of the formation of the peptide cross-links in the
formation of the cell wall of
Staphylcoccus
aureus
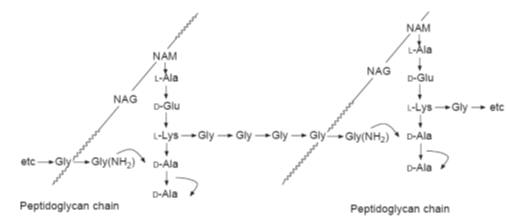
The
b-lactam group of antibiotics inhibit cell wall
synthesis by inhibiting the transpeptidases responsible for the cross-linking
between the peptidoglycan chains. This group of antibiotics, named after the
b-lactam ring that they all have in common,
includes the widely used penicillins and the cephalosporins (Fig.). Both of
these groups of
beta-lactam antibiotics are
more effective against Gram-positive bacteria than Gram-negative bacteria.
However, some cephalosporins, such as ceftazidime, which is administered
intravenously, are very effective against Gram-negative bacteria.
Examples of the range of penicillins and cephalosporins. The R residues of
ampicillin, amoxicillin and ceftazidime have D configurations
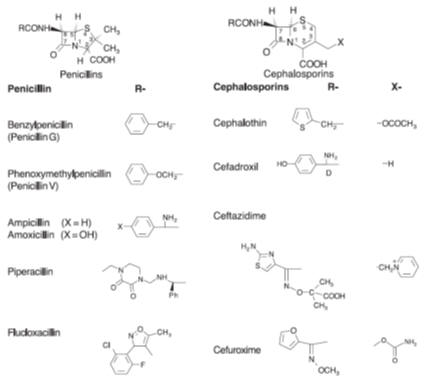
The
b-lactam antibiotics have to reach the plasma
membrane of the bacteria before they can act. As the outer surfaces of
Gram-positive bacteria are covered by a thin layer of teichoic acids. they offer
less resistance to drug penetration than Gram-negative bacteria, where the drug
has to penetrate both the outer membrane and the periplasmic space (Fig.) before
it can interfere with cell wall synthesis. In Gram-negative bacteria the drug
diffuses through the outer membrane via
porin channels
formed by integral trimeric proteins. A large
number of these channels, with diameters of about 1.2 nm, are found in each
bacterial cell wall. However, not all porin channels are able to transport
b-lactam drugs. Some bacterial genera such as
Pseudomonas
are resistant to
b-lactam
antibiotics because their porin channels will not allow the transport of these
drugs.
Once through the
outer membrane the drug diffuses across the periplasmic space, which contains
b-lactamases that can inactivate the drug.
Gram-positive bacteria also produce these enzymes, which they release into the
extracellular fluid. Finally, the drug penetrates to the outer surface of the
plasma membrane where it binds to, and blocks the action of, the transpeptidases
and other proteins involved in cell wall synthesis. The precise nature of the
blocking mechanism has not yet been fully elucidated but appears to involve the
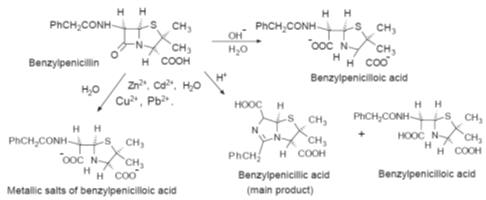
b-lactam ring system. This ring system is very
reactive and is easily decomposed by acid and base catalysed hydrolysis (Fig.),
the rate of which depends on the structure of the penicillin.
Some of the decomposition routes of the
b-lactam
ring of benzylpenicillin
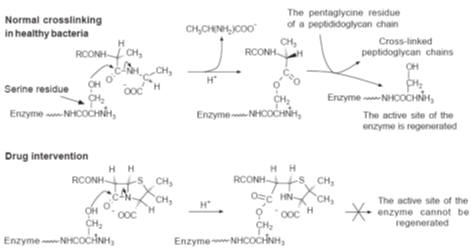
The final stage
in the formation of the cross-links between the peptidoglycan chains in bacteria
is catalysed by a glycopeptide transpeptidase. It is believed that the hydroxyl
group of a serine residue in this enzyme displaces the last alanine residue from
the tetrapeptide chain. The displaced alanine diffuses away from the reaction
site, allowing attack by the amino group of the terminal glycine of the
pentaglycine chain on the alanine bound to the enzyme to complete the peptide
linkage and regenerate the enzyme (Fig.). However, it is thought that since the
geometry of the penicillins resembles that of the alanyl-alanyl unit the
bacteria mistakes the drug for its normal substrate. The
b-lactam ring reacts with the enzyme to form a
covalently bound acyl derivative. The 1,3-thiazolidine ring of this derivative
is believed to prevent a pentaglycine unit from attacking the enzyme–acyl
linkage and regenerating the active site of the enzyme.
A schematic outline of the chemistry proposed for the action of penicillins
Penicillins are
unstable under acid conditions, the rate of decomposition depending on which
penicillin is being considered. For example, piperacillin (Fig.) is so acid
labile that it has to be administered by intravenous infusion. This means that
the acid conditions of the stomach could reduce the amount of the drug reaching
the general circulatory system and hence its effectiveness. The reactivity of
penicillins under acid conditions can be attributed to the presence of a
reactive four-membered lactam ring, the carbonyl group of which is readily
attacked by nucleophiles under acid conditions.
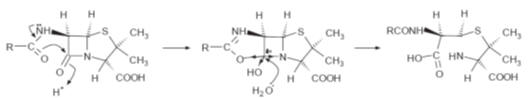
This reactivity
is believed to be enhanced by the presence of the acyl side chain in a so-called
neighbouring group effect.
This group is believed to enhance the electronegative nature of the oxygen of
the carbonyl group of the lactam, which makes the carbon of the lactam group
more susceptible to nucleophilic attack (Fig.).
A possible mechanism for the enhancement of the reactivity of the carbonyl group
of the lactam by a neighbouring group
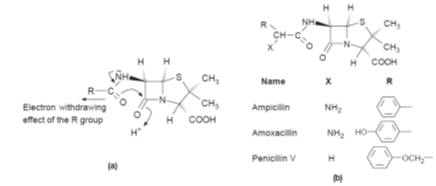
Consequently,
some acid-resistant penecillins have been produced by introducing an electron
withdrawing group on the alpha carbon of the side chain. This reduces
neighbouring group participation and, as a result, the reactivity on the
lactam’s carbonyl group (Fig.). This approach led to the development of
penicillin V (phenoxymethyl-penicillin), amoxacillin and ampicillin.
Cephalosporins
usually exhibit a greater resistance to acid hydrolysis than penicillins.
However, the first generation of cephalosporins were not as potent as the
penicillins but were
active against a
wider range of bacteria. However, their absorption from the GI tract is often
poor and so they
have to be given by injection. Consequently, cephalosporin C, first isolated by
workers in Oxford University in the late 1940s, was used as the lead to develop
more active
analogues (Fig.).
A large number of different cephalosporins are now in clinical use. The
relationship between the structures of
b-lactams
and their activity has been the subject of much discussion. Originally it was
believed that the amide-linked side chain, the carboxylic acid at position 2 and
the fused thio-ring systems were all essential for the pharmacological activity
of
b-lactam antibiotics. However, the
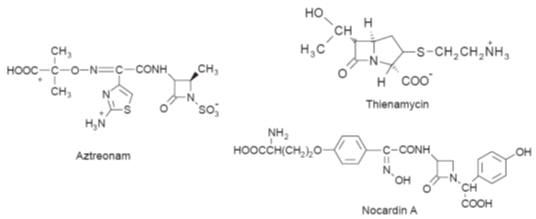
discovery of
b-lactams such as thienamycin,
aztreonam and nocardin A (Fig.), whose structures do not contain all these
functional groups, suggests that the
b-lactam
ring is the only essential requirement for activity.
(a)
It is believed that the electron withdrawing effect of the R group reduces the
ability of the electrons of the carbonyl group of the amide link to influence
those of the carbonyl group of the lactam. (b)
Examples of penicillins in clinical use with an electron withdrawing R group
The increasing
number of bacteria resistant to
b-lactam
antibiotics is becoming a major problem. Resistance to penicillins and
cephalosporins by some bacteria is mainly due to inactivation of the drug by
hydrolysis of the lactam ring catalysed by the
b-lactamases
produced by that bacterium. However, in general, penicillins tend to be more
susceptible than cephalosporins to hydrolysis catalysed by
b-lactamases.
Both
Gram-positive and Gram-negative bacteria produce
b-lactamases.
In the former case the enzyme is liberated into the medium surrounding the
bacteria. This results in inactivation of the penicillin, cephalosporin and
other
b-lactam drugs before the drug
reaches the bacteria. However, with Gram-negative bacteria, the hydrolysis takes
place within the periplasmic space. In addition some Gram-negative bacteria
produce
Acylases
which can cleave the side chains of penicillins. Bacteria that have
developed a resistance to
b-lactam
antibiotics are often treated using a dosage form incorporating a
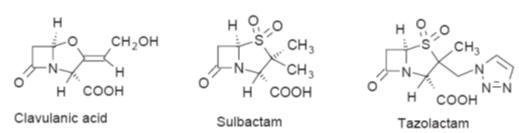
b-lactamase inhibitor such as clavulanic acid,
sulbactam or tazobactam (Fig.) and Table.
Examples of
b-lactam antibiotics that do
not contain a thiol-ring system
Examples of
b-lactamase inhibitors used
in penicillin dosage forms
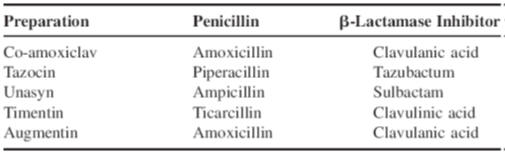
Examples of dosage forms containing
b-lactamase
inhibitors

An alternative
approach to the problem of bacterial resistance was the development of
b-lactamase-resistant drugs. The strategy
adopted by Beecham Research Laboratories for penicillins in 1961 was to use
bulky substituents close to the labile lactam ring in order to use
steric hindrance
to prevent the drug binding to the enzyme’s active site and undergoing lactam
hydrolysis. This approach resulted in the discovery of methicillin. However,
methicillin had a reduced potency compared with other penicillins and was acid
labile to the extent that it could only be administered by injection. Further
work by Beecham using an isoxazole ring in the side chain resulted in the
discovery of flucloxacillin (Fig.), oxacillin, cloxacillin and dicloxacillin.
These drugs are used against Gram positive bacteria. They are inactive against
Gram negative bacteria. However, oxacillin is also acid resistant.
The introduction
of a bulky
syn
a-oximino
group (-C-N-O-) side chain in so-called ‘second-generation cephalosporins’
improved their stability towards
b-lactamases
and esterases by probably sterically hindering the hydrolysis of the lactam. For
example, cefuroxime (Fig.) is active against a wide range of Gram-positive and
Gram-negative bacteria. Further development has resulted in the discovery of
so-called ‘third- and fourth-generation’ cephalosporins that also exhibit
enhanced resistance to
b-lactamases. This increased
resistance is believed to be partly due to the presence of a
syn
a-oximino side chain. In the
third-generation cephalosporins, such as ceftazidine (Fig.), the presence of an
aminothiazole group is thought to increase the ease of transfer of the drug
through the outer membrane of Gram-negative bacteria, which would account for
their good Gram-negative activity and variable Gram-positive activity. The
fourth-generation cephalosporins, such as cefepime and cefpirome, are
zwitterions. They have a high potency against Gram-positive, Gram-negative and
Pseudomonas aeruginosa.

Polypeptide antibiotics
A large number of polypeptide antibiotics have
been discovered. They are active against a wide variety of microorganisms and
operate by a range of mechanisms. Vancomycin and teicoplanin (teichomycin A2)
act by inhibiting the cross-linking of the peptidoglycan chains in bacterial
cell walls. Other polypeptide antibiotics such as gramicidin and viomycin, which
is used to treat TB, have different mechanisms of action. Vancomycin (Fig.a) is
a glycopeptide antibiotic that was isolated from
Streptomyces orientalis
in 1955 and inhibits the formation of the
peptide links between the peptidoglycan chains. In spite of the extensive use of
the drug, very little bacterial resistance to vancomycin has developed. It is
mainly used for Gram-positive infections but is irritating on intravenous
injection. Oral administration does not give useful blood levels. However, the
drug is used orally to treat pseudomembranous enterocolitis caused by high
concentrations of
Clostridium difficile
in the intestine.
The structure of
vancomycin (Fig.a) is based on a tricyclic ring system of aliphatic and aromatic
amino acids with a disaccharide side chain. This structure is rigid with a
peptide-lined pocket that has a strong affinity for D-ala-D-ala residues.
Vancomycin inhibits cell wall synthesis by binding to the D-ala-D-ala end group
of the pentapeptide chain of the peptidoglycan cell wall precursor. NMR
spectroscopy and molecular modelling suggest that the D-ala-D-ala residue is
multiple hydrogen-bonded to the vancomycin in this pocket. This inhibits the
formation of the peptide cross-links between the polyglycan chains (Fig.), which
results in a loss of bacterial cell wall integrity.


Those bacteria
that exhibit resistance to the drug appear to have replaced the D-ala-D-ala unit
of the pentapeptide residue by a D-ala-D-lactate unit. This structural change is
believed to reduce the number of hydrogen bonds between the drug and the
D-ala-D-lactate of the peptidoglycan precursor by one. However, this small
change is sufficient to prevent the drug operating efficiently. Teicoplanin is a
complex of five compounds (A2-1, A2-2 to A2-5) with similar structures (Fig. b)
that is active against a range of Gram-positive and Gram-negative bacteria.
It was isolated
in 1976 from
Actinoplanes teichomycetius.
The components of the mixture were separated in 1983 and their structures
elucidated in 1984. Teicoplanin has good water solubility but significantly
better lipid solubility than vancomycin. It is generally less toxic than
vancomycin and its long half-life means that it need only be administered once a
day. Its mechanism of action is believed to be identical to that of vancomycin.
Surfactants used as antibacterial agents

Surface active
agents disrupt cell membranes because they dissolve in both the aqueous
extracellular fluid and the lipid membrane. This lowers the surface tension of
the membrane, which allows water to flow into the cell and ultimately results in
lysis and bactericidal action. In all cases a balance between the hydrophilic
and lipophilic sections of the molecule is essential for action. Both cationic
and non-ionic surfactants are used (Fig). In addition, detergent surfactants,
such as sodium dodecyl sulphate, are also used to remove proteins from cell
membranes.
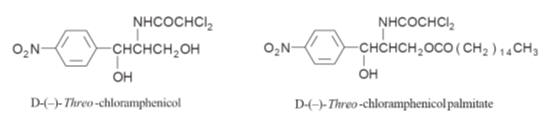
Chloramphenicol
Chloramphenicol
was first isolated by Ehrlich
et al.
in 1947 from the microorganism
Streptomyces venezuelae,
which was found in a soil sample from Venezuela. It is a broad spectrum
antibiotic whose structure contains two asymmetric centres. However, only the
D-(-)-threo
form is active. Its use can cause serious side effects and so it is
recommended that chloramphenicol is only used for specific infections. It is
often administered as its palmitate in order to mask its bitter taste. The free
drug is liberated from this ester by esterase-catalysed hydrolysis in the
duodenum. Chloramphenicol has a poor water solubility (2.5 g
dm-3)
and so it is sometimes administered in the form of its more soluble sodium
hemisuccinate salt, which acts as a prodrug. Chloramphenicol is believed to act
by inhibiting the elongation stage in protein synthesis in prokaryotic cells. It
binds reversibly to the 50S ribosome subunit and is thought to prevent the
binding of the aminoacyl–tRNA complex to the ribosome. However, its precise mode
of action is not understood.
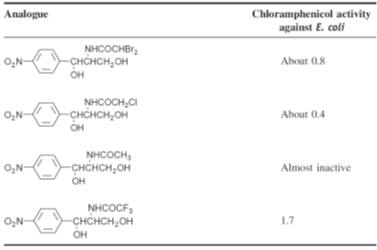
Investigation of
the activity of analogues of chloramphenicol showed that activity requires a
para-electron
withdrawing group. However, substituting the nitro group with other electron
withdrawing groups gave compounds with a reduced activity. Furthermore,
modification of the side chain, with the exception of the difluoro derivative,
gave compounds that had a lower activity than chloramphenicol (Table). These
observations suggest that D-(-)-threo-chloramphenicol
has the optimum structure of those tested for activity.
The activity against
E. coli
of some analogues of
chloramphenicol relative to chloramphenicol
The synthesis of
chloramphenicol was first reported by Controulis J
et al.
in 1949 (Fig.). Numerous synthetic routes have since been devised
for the synthesis of chloramphenicol, the commercial routes usually starting
with 4-nitroacetophenone. Chloramphenicol is now manufactured by both totally
synthetic and microbiological routes.
Antiviral drugs
It has been found
that viruses utilise a number of virus-specific enzymes during replication.
These enzymes and the processes they control are significantly different from
those of the host cell to make them a useful target for medicinal chemists.
Consequently, antiviral drugs normally act by inhibiting viral nucleic acid
synthesis, inhibiting attachment to and penetration of the host cell or
inhibiting viral protein synthesis.
An outline of the mechanism for DNA chain termination used by some antiviral
drugs. The
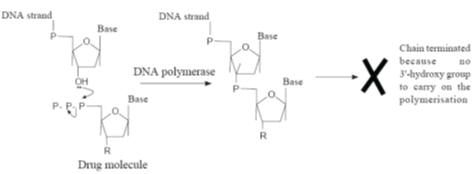
Nucleic acid synthesis inhibitors
Nucleic acid
synthesis inhibitors usually act by inhibiting the polymerases or reverse
transcriptases required for nucleic acid chain formation. However, because they
are usually analogues of the purine and pyrimidine bases found in the viral
nucleic acids, they are often incorporated into the growing nucleic acid chain.
In this case their general mode of action frequently involves conversion to the
corresponding 5’-triphosphate
by the host cell’s cellular kinases. This conversion may also involve specific
viral enzymes in the initial monophosphorylation step. These triphosphate drug
derivatives are incorporated into the nucleic acid chain where they terminate
its formation. Termination occurs because the drug residues do not have the 30-hydroxy
group necessary for the phosphate ester formation required for further growth of
the nucleic acid chain. This effectively inhibits the polymerases and
transcriptases that catalyse the growth of the nucleic acid (Fig.).
It is not
possible to list all the known antiviral agents in this text so only a
representative selection are discussed.
Aciclovir
Aciclovir was the
first effective antiviral drug. It is effective against a number of herpes
viruses, notably simplex, varicella-zoster (shingles), varicella (chickenpox)
and Epstein–Barr virus (glandular fever). It may be administered orally and by
intravenous injection as well as topically. Orally administered doses have a low
bioavailability.
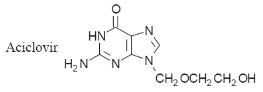
The action of
aciclovir is more effective in virus-infected host cells because the viral
thymidine kinase is a more efficient catalyst for the monophosphorylation of
aciclovir than the thymidine kinases of the host cell. This leads to an increase
in the concentration of the aciclovir triphosphate, which has 100-fold greater
affinity for viral DNA polymerase than human DNA polymerase. As a result, it
preferentially competitively inhibits viral DNA polymerase and so prevents the
virus from replicating. However, resistance has been reported due to changes in
the viral mRNA responsible for the production of the viral thymidine kinase.
Aciclovir also acts by terminating chain formation. The aciclovir–DNA complex
formed by the drug also irreversibly inhibits DNA polymerase.
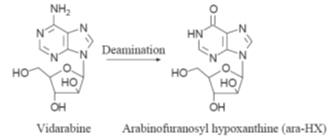
Vidarabine
Vidarabine is
active against herpes simplex and herpes varicella-zoster. However, the drug
does give rise to nausea, vomiting, tremors, dizziness and seizures. In addition
it has been reported to be mutagenic, teratogenic and carcinogenic in animal
studies. Vidarabine is administered by intravenous infusion and topical
application. It has a half-life of about one hour, the drug being rapidly
deaminated to arabinofuranosyl hypoxanthine (ara-HX) by adenosine deaminase.
This enzyme is found in the serum and red blood cells. Ara-HX, which also
exhibits a weak antiviral action, has a half-life of about 3.5 hours.
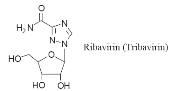
Ribavirin
Ribavirin is
effectively a guanosine analogue. It is active against a wide varietyof DNA and
RNAviruses but the mechanism by which it acts is not understood. It is mainly
used in aerosol form to treat influenza and other respiratory viral infections.
Intravenous administration in the first 6 days of onset has been effective in
reducing deaths from Lassa fever to 9 per cent. Ribavirin has also been shown to
delay the onset of full-blown AIDS in patients with early symptoms of HIV
infection. However, administration of the drug has been reported to give rise to
nausea, vomiting, diarrhoea, deterioration of respiratory function, anaemia,
headaches and abdominal pain. The mechanism by which it acts may differ from one
virus to another.
Zidovudine (AZT)
Zidovudine was
originally synthesised in 1964 as an analogue of thymine by J. Horwitz as a
potential antileukaemia drug. It was found to be unsuitable for use in this role
and for 20 years was ignored, even though in 1974W. Osterag
et al. reported that it was active against Friend leukaemia virus, a
retrovirus. However, the identification in 1983 of the retrovirus HIVas the
source of AIDS resulted in the virologist M. St Clair setting up a screening
programme for drugs that could attack HIV. Fourteen compounds were selected and
screened against Friend leukaemia virus and a second retrovirus called Harvey
sarcoma virus. This screen led to the discovery of zidovudine (AZT), which was
rapidly developed into clinical use on selected patients in 1986.
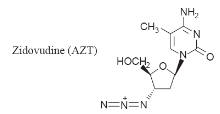
AZT is converted
by the action of cellular thymidine kinase to the 50-triphosphate.
This inhibits the enzyme reverse transcriptase in the retrovirus, which
effectively prevents it from forming the viral DNA necessary for viral
replication. The incorporation of AZT into the nucleic acid chain also results
in chain termination because the presence of the 30-azide
group prevents the reaction of the chain with the 50-triphosphate
of the next nucleotide waiting to join the chain (Fig.). AZT is also active
against mammalian DNA polymerase and although its affinity for this enzyme is
about 100-fold less this action is thought to be the cause of some of its
unwanted side effects.
Zidovudine is
active against the retroviruses that cause AIDS (HIV virus) and certain types of
leukaemia. It also inhibits cellular
a-DNA
polymerase but only at concentrations in excess of 100-fold greater than those
needed to treat the viral infection. The drug may be administered orally or by
intravenous infusion. The bioavailability from oral administration is good, the
drug being distributed into most body fluids and tissues. However, when used to
treat AIDS it has given rise to gastrointestinal disorders, skin rashes,
insomnia, anaemia, fever, headaches, depression and other unwanted effects.
Resistance increases with time. This is known to be due to the virus developing
mutations’ which result in changes in the amino acid sequences in the reverse
transcriptase.
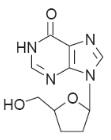
Didanosine
Didanosine is
used to treat some AZT-resistant strains of HIV. It is also used in combination
with AZT to treat HIV. Didanosine is administered orally in dosage forms that
contain antacid buffers to prevent conversion by the stomach acids to
hypoxanthine. However, in spite of the use of buffers the bioavailability from
oral administration is low. The drug can cause nausea, abdominal pain and
peripheral neuropathy, amongst other symptoms. Drug resistance occurs after
prolonged use. Didanosine is converted by viral and cellular kinases to the
monophosphate and then to the triphosphate. In this form it inhibits reverse
transcriptase and in addition its incorporation into the DNA chain terminates
the chain because the drug has no 30-hydroxy group (Fig.).
Host cell penetration inhibitors
The principal
drugs that act in this manner are amantadine and rimantadine (Fig.). Both
amantadine and rimantadine are also used to treat Parkinson’s disease. However,
their mode of action in this disease is different from their action as antiviral
agents.
Examples of host cell penetration inhibitors
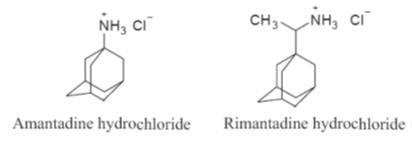
Amantadine hydrochloride
Amantadine
hydrochloride is effective against influenza A virus but is not effective
against the influenza B virus. When used as a prophylactic, it is believed to
give up to 80 per cent protection against influenza A virus infections. The drug
acts by blocking an ion channel in the virus membrane formed by the viral
proteinM2. This is believed to inhibit the disassembly of the core of the virion
and its penetration of the host. Amantadine hydrochloride has a good
bioavailability on oral administration, being readily absorbed and distributed
to most body fluids and tissues. Its elimination time is 12–18 hours. However,
its use can result in depression, dizziness, insomnia and gastrointestinal
disturbances, amongst other unwanted side effects.
Rimantadine hydrochloride
Rimantadine
hydrochloride is an analogue of amantadine hydrochloride. It is more effective
against influenza A virus than amantadine. Its mode of action is probably
similar to that of amantadine. The drug is readily absorbed when administered
orally but undergoes extensive first-pass metabolism. However, in spite of this,
its elimination half-life is double that of amantadine. Furthermore, CNS side
effects are significantly reduced.
Inhibitors of viral protein synthesis
The principal
compounds that act as inhibitors of protein synthesis are the
interferons.
These compounds are members of a naturally occurring family of glycoprotein
hormones (RMM 20 000–160 000), which are produced by nearly all types of
eukaryotic cell. Three general classes of interferons are known to occur
naturally in mammals, namely:
the
a-interferons produced by leucocytes,
b-interferons produced by fibroblasts and
g-interferons produced by T lymphocytes. At
least twenty
a-, two
b- and two
g-interferons
have been identified. Interferons form part of the human immune system. It is
believed that the presence of virions, bacteria and other antigens in the body
switches on the mRNA that controls the production and release of interferon.
This release stimulates other cells to produce and release more interferon.
Interferons are thought to act by initiating the production in the cell of
proteins that protect the cells from viral attack. The main action of these
proteins takes the form of inhibiting the synthesis of viral mRNA and viral
protein synthesis.
a-Interferons also enhance the
activity of killer T cells associated with the immune system.
A number of
a-interferons have been manufactured (see Table
10.10 on page 394) and proven to be reasonably effective against a number of
viruses and cancers. Interferons are usually given by intravenous, intramuscular
or subcutaneous injection. However, their administration can cause adverse
effects, such as headaches, fevers and bone marrow depression, that are dose
related.
The formation and
release of interferon by viral and other pathological stimulation has resulted
in a search for chemical inducers of endogenous interferon. Administration of a
wide range of compounds has resulted in the induction of interferon production.
However, no clinically useful compounds have been found for humans’ although
tilorone is effective in inducing interferon in mice.
Antimalarial compounds
The
causative agents of malaria are plasmodia, unicellular organisms (Order
Hemosporidia, Class Protozoa). The infective form, the sporozoite, is inoculated
into skin capillaries when infected female
Anopheles
mosquitoes (A) suck blood from humans. The sporozoites
invade liver parenchymal cells, where they develop into primary tissue
schizonts. These give rise to numerous merozoites that enter the blood. The
preerythrocytic stage is asymptomatic. In blood, the parasite enters
erythrocytes (erythrocytic stage), where it again multiplies by schizogony,
resulting in the formation of more merozoites. Rupture of the infected
erythrocytes releases the merozoites and pyrogens. A fever attack ensues and
more erythrocytes are infected. The generation period for the next crop of
merozoites determines the interval between fever attacks. With
Plasmodium vivax
and
P. ovale,
there can be a parallel multiplication in the liver (paraerythrocytic stage).
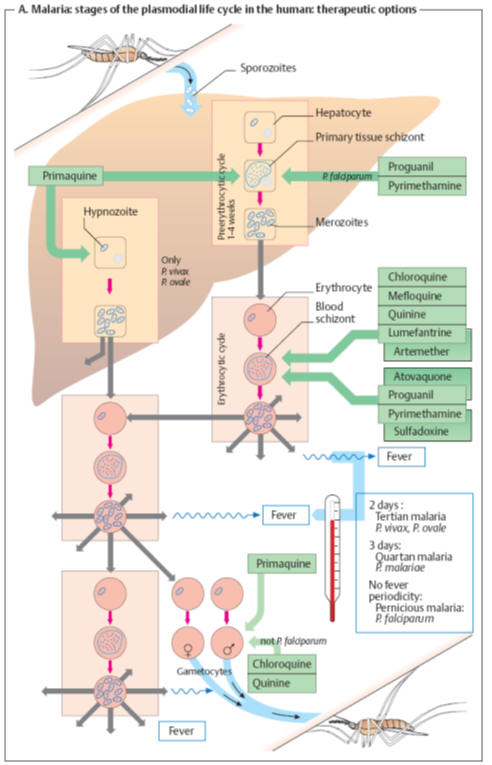
Moreover,
some sporozoites may become dormant in the liver as “hypnozoites” before
entering schizogony. Different
antimalarials
selectively kill
the parasite’s different developmental forms. The mechanism of action is known
for some agents:
Chloroquine
and
quinine
accumulate within the acidic vacuoles of blood schizonts and inhibit
polymerization of heme released from digested hemoglobin, free heme being toxic
for the schizonts.
Pyrimethamine
inhibits protozoal dihydrofolate reductase, as does
chlorguanide
(proguanil)
via its active metabolite cycloguanil. The sulfonamide
sulfadoxine
inhibits
synthesis of dihydrofolic acid. Dihydrofolate reductase is also blocked by
cycloguanil, the active form of
proguanil.
Atoquavone
suppresses synthesis of pyrimidine bases, probably by interfering with
mitochondrial electron transport.
Artemesinin derivatives
(artemether, artesunate) originate from the East Asian plant Qinghaosu (Artemisia
sp.) Its
antischizontal effect appears to involve a reaction between heme iron and the
epoxide group of these compounds. Antimalarial drug choice takes tolerability
and plasmodial resistance into account.
Tolerability
The
oldest antimalarial, quinine, has the smallest therapeutic margin. All newer
agents are rather well tolerated.
Plasmodium falciparum, responsible for the most dangerous form
of malaria, is particularly prone to develop
drug resistance. The prevalence of resistant strains rises
with increasing frequency of drug use. Resistance has been reported for
chloroquine and also the combination pyrimethamine/sulfadoxine.
Drug choice for antimalarial chemoprophylaxis
In areas
with a risk of malaria, continuous intake of antimalarials affords the best
protection against the disease, though not against infection. Primaquine would
be effective against primary tissue schizonts of all plasmodial species;
however, it is not used for long-term prophylaxis because of unsatisfactory
tolerability and the risk of plasmodial resistance. Instead, prophylactic
regimens employ agents against blood schizonts. Depending on the presence of
resistant strains, use can be made of chloroquine, and/or proguanil, mefloquine,
the tetracycline doxycycline, as well as the combination of atoquavone and
proguanil. These drugs do not prevent the (symptom-free) hepatic infection but
only the disease-causing infection of erythrocytes (“suppression therapy”). On a
person’s return from an endemic malaria region, a two-week course of primaquine
is adequate for eradication of the late hepatic stages (P.
vivax
and
P.
ovale). Protection frommosquito bites (with
nets, skin-covering clothes, etc.) is a very important prophylactic measure.
Therapy
Antimalarial therapy employs the same agents, in addition to the combinations of
artemether plus lumefantrine or pyrimethamine plus sulfadoxine.
Enzymes and drug resistance
Drug resistance
occurs when a drug no longer has the desired clinical effect. This may be due to
either a natural inbuilt resistance in some individuals and organisms or may
arise naturally in the course of treatment. The former is probably due to
differences in the genetic code of individuals within a species whilst the
latter arises because of natural selection. In natural selection the drug kills
the susceptible strains of an organism but does not affect other strains of the
same organism. Consequently, these immune strains multiply and become the common
strain of the organism, which subsequently results in ineffective drug
treatment. Resistance occurs on an individual basis and so is not usually
detected until a wide sample of the population has been treated with or
indirectly exposed to the drug. Its detection necessitates the discovery of new
drugs to treat the condition. Its emergence is probably due to the widespread
and poorly controlled use of a drug. For example, the generous use of
antibiotics in farming is strongly suspected to be the reason for an increase in
antibiotic-resistant strains of bacteria in humans. The response of medicinal
chemists to resistance is either to devise new drugs or to modify existing
drugs. This approach suffers from the high probability of being unsuccessful as
well as time consuming and expensive. In the light of human experience it would
be better if, in future, we reduced the possibility of resistance by using the
effective existing drugs more intelligently. Drug resistance can be linked to a
change in either the permeability of the membranes of the organism or an enzyme
system(s) of the organism. Enzymes may be involved in drug resistance in a
number of ways but in many cases resistance may be due to several different
processes occurring at approximately the same time.
Changes in enzyme concentration
A significant
increase or decrease from the normal concentration of an enzyme can result in
resistance to a drug. The overproduction of an enzyme can have two effects:
1. The target
process catalysed by the enzyme will not be inhibited because excess enzyme is
produced. For example, the resistance of malarial parasites is believed to be
caused by overproduction of dihydrofolate reductase due to the drug stimulating
the parasite’s RNA.
2. The increased
production of enzymes that inactivate the drug, for example
beta-lactamases inactivate most penicillins and
cephalosporins by hydrolysing their
b-lactam
rings. A number of enzymes deactivate inhibitors by incorporating (conjugation)
phosphate by phosphorylation of hydroxyl groups, adenine by adenylation of
hydroxyl groups or acetyl by acetylation of amino groups in the inhibitor’s
structure (Fig.). ATP is believed to be the usual provider of phosphate and
adenylic acid, whilst acetyl coenzyme A is thought to be the normal source of
acetyl groups. For example, resistance to the antibiotic kanamycin A can occur
by all three routes (Fig.) although kanamycin A-resistant bacteria do not
normally use all three routes.
The inhibition of kanamycin A by enzymatic inactivation. The arrows indicate the
structure and position of the result of an enzyme reaction

Many
aminoglycoside antibiotics are susceptible to this type of enzyme inhibition.
However, amikacin (AK) where the C1-NH2 of kanamycin A has been acylated by
S-a-hydroxy-gamma-aminobutanoic acid is not susceptible to 3’-phosphorylation
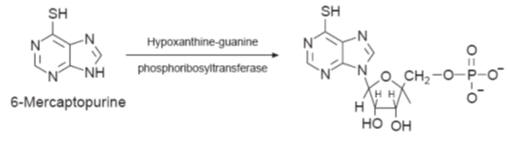
and 2”-adenylation. The underproduction of an enzyme
could result in insufficient enzyme being present to produce the active form of
a drug from a prodrug. For example the resistance to the antileukaemia drug
6-mercaptopurine is caused by a reduced production of hypoxanthine–guanine
phosphoribosyltransferase, the enzyme required to convert the prodrug to its
active ribosyl 5’-monophosphate
derivative.
These changes in
the production of the enzyme are believed to be due to genetic changes in the
organism.
An increase in the production of the substrate
Increased
production of the substrate can prevent competitive reversible inhibitors from
binding to the active site in sufficient quantities to be effective. The high
concentration of the substrate moves the position of equilibrium to favour the
formation of the E–S complex.
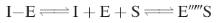
For example, the
inhibition of dihydropteroate synthetase by sulphonamides results in a build-up
of
p-aminobenzoic acid. This increase in substrate
concentration prevents sulphonamides from inhibiting dihydropteroate synthetase,
which is a key enzyme in the production of the RNA necessary for bacterial
reproduction. Similarly, a build-up of angiotensin I will overcome the effect of
ACE inhibitors. It is thought to be the reason for the concentration of plasma
angiotensin II returning to normal in some cases where there has been a chronic
administration of ACE inhibitors.
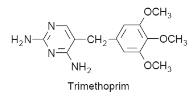
Changes in the structure of the enzyme
Changes in the
structure of the target enzyme result in a structure that is not significantly
inhibited by the drug. However, the modified enzyme is still able to produce the
normal product of the reaction, which allows the unwanted metabolic pathway to
continue to function. For example, resistance to the antibiotic trimethoprim is
believed to be due to a plasmid-directed change in the structure of
dihydrofolate reductase in the bacteria.
Similarly, the
resistance of the
E. coli
strains to sulphonamides
has been shown to be due to their containing a sulphonamide-resistant
dihydropteroate synthase.
The use of an alternative metabolic pathway
The blocking of a
metabolic pathway by a drug can result in the opening of a new pathway
controlled by a different enzyme that is not inhibited by the same drug.
Agonist: A drug that mimics
the endogenous receptor ligand to activate the receptor to produce a biological
response is called as an agonist. Several agonists are able to produce the
target maximum response without completely occupying all the receptors.
Partial agonist: A drug that binds and activates a
receptor but does not elicit a full response is known as a partial agonist. A
partial agonist can block the effect of a full agonist. In the presence of high
concentrations of a partial agonist, the action of a full agonist can be reduced
to the maximum response elicited by the partial agonist. However, the intrinsic
activity would be greater than zero but less than 1 that of a full agonist.
Inverse agonist: An inverse agonist is a molecule
or agent that binds to the same receptor site as an agonist and is considered to
be a full agonist. However, it exerts the opposite pharmacological response to
that of a normal agonist, i.e. demonstrates negative efficacy. Constitutive
activity refers to the ability of a receptor in producing its biological
response in the absence of a bound ligand. The constitutive activity of a
receptor may be blocked by an inverse agonist.
Biased agonist: G protein-coupled receptors
(GPCRs) are capable of signalling with different efficacies to their multiple
downstream pathways, a phenomenon referred to as biased agonism. Biased agonism
is one of the fastest growing areas in GPCR pharmacology. Biased agonism has
been primarily reported as a phenomenon of synthetic ligands and the biological
importance of such signalling is unclear.
Antagonist: A drug that binds to a
receptor but does not elicit a response is referred to as an antagonist.
Importantly, the antagonist must block the action of the agonist at the receptor
site. Antagonist can shift the concentration–response curve of an agonist to the
right by reducing its fractional occupancy. High concentrations of the
antagonist may block the actions of the agonist completely. However, antagonists
have no intrinsic activity and therefore they do not produce any effects. There
are two main types of antagonists. Competitive antagonists compete with the
agonist for same receptor binding site, but the binding is reversible. It shifts
the concentration-response curve of the agonist to the right without any
reduction in maximal response. Non-competitive antagonists bind irreversibly to
a receptor site and thereby reduce the ability of an agonist to bind and produce
a response. The non-competitive antagonism is a slow process which resulting in
a prolonged antagonistic effect.
Drug affinity and efficacy
Affinity can be defined as
the extent or fraction to which a drug binds to receptors at any given drug
concentration or the firmness with which the drug binds to the receptor. The
mathematical model of affinity of a drug for the receptor was first described by
Irving Langmuir Kenakin (2004). Affinity is one of the factors that determine
potency. Affinity is inversely proportional to the potency of a drug
(1Kd),
where Kd is the dissociation constant. The strength of the binding (interaction)
of a ligand and its receptor can be described by affinity. The higher the Kd
value, the weaker the binding and the lower the affinity. The opposite occurs
when a drug has a low Kd. Potency is a measure of necessary amount of the drug
to produce an effect of a given magnitude. In general, potency is denoted as the
median effective concentration/dose as EC50/ED50/Kd.
Efficacy (intrinsic activity)
is the ability of a drug to illicit a pharmacological response (physiological)
when interaction occurs with a receptor (relationship between response and
occupancy of receptor). Efficacy depends on the efficiency of the receptor
activation to cellular responses and the formation of number of drug-receptor
complexes.
•Full agonists: efficacy = 1.
•Partial agonists: efficacy > 0 and < 1.
•Competitive antagonists: efficacy = 0.
Clark’s occupancy theory
Clark in the
1920s visualised the drug–receptor interaction as being a bimolecular dynamic
equilibrium with
the drug molecules continuously binding to and leaving the receptor, that is:
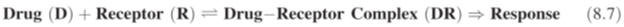
receptors
occupied by the drug: the greater the number occupied, the greater the
pharmacological
effect, that is:
![]()
According to
Clark a maximum response would be obtained when all the receptors were
occupied, that
is:
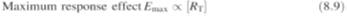
where
RT
is the total number of receptors. It follows from equations (8.8) and (8.9) that
for
a given dose of a
drug the fraction of the maximum response is given by:
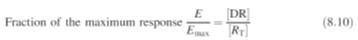
The dissociation
of the drug–receptor complex may be represented as:

and applying the
law of mass action:

where
KD
is the dissociation constant for the drug–receptor complex.
But the total
receptor concentration is:

Substituting
equation (8.13) in equation (8.12):

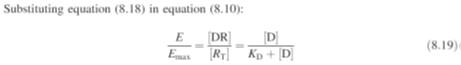
Equation (8.19)
shows that the relationship between
E
and molar drug concentration [D] is in the form of a rectangular hyperbola,
whilst that between
E
and log [D] is
sigmoidal. These theoretical relationships derived using Clarks’ theory are
often in good agreement with the experimental results (Fig.) obtained in a
number of investigations. Furthermore, substituting the value of
E/Emax
=
1/2 in equation (8.19) gives the relationship:

where EC50 is the
molar concentration of the drug that produces half the maximum biological
response observed when a ligand binds to a receptor. However, in practice this
theoretical relationship does appear to be the exception rather than the rule.
The value of the dissociation constant
KD is a measure of
the affinity of the drug for the receptor. Drugs with small
KD
values have a large affinity for the receptor whilst those with high values have
a low affinity. As a result,
KD
values are used to compare the activities of a series of analogues during drug
development. The value of
KD may be
determined experimentally from tissue binding experiments using a radioactive
form of the drug. The data obtained from this type of experimental work may be
analysed using a Scatchard plot of the ratio of bound to free ligand against
bound drug (Fig.). This gives a straight line with a slope of
-1/KD
provided that the drug binds to only one type of receptor. In industry the data
are now analysed by the use of a computerised method of least squares.
The correlation of experimental results and those predicted using Clark’s theory
for the stimulated contraction of guinea-pig ileum by acetylcholine
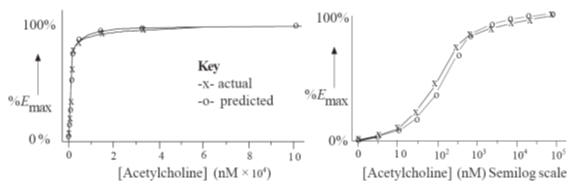
Although Clark’s
occupancy theory is still a cornerstone of pharmacodynamics, a number of its
assumptions have now been shown to be incorrect. It is now known that:
·
the formation of many drug–receptor complexes is not reversible;
·
the receptor sites are not always independent;
·
the formation of the complex may not be bimolecular: for example, two
acetylcholine
molecules bind to nACh receptors of ion
channels;
·
a maximum response may be obtained before all the receptors are occupied;
·
the response is not linearly related to the proportion of receptors occupied,
especially
in the case of
partial agonists.
In the 1950s.
Ariens and Stephenson separately modified Clark’s theory to account for the
existence of agonists, partial agonists and antagonists. They based their
modifications on a proposal by Langley in 1905, which visualised the action of a
receptor as taking place in two stages. The first stage was the binding of the
ligand to the receptor, which was controlled by the ligand’s affinity for the
receptor. The second stage was the initiation of the biological response. Ariens
said that this second step was governed by the ability of the ligand–receptor
complex to initiate a response. Ariens called this ability the
intrinsic activity
(a), whilst
Stephenson referred to it as the
efficacy
(e)
of the ligand–receptor complex. Intrinsic activity may be defined as:
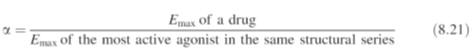
Using the concept
of intrinsic activity Clark’s equation (equation 8.19) becomes:

When
a
=
1 for ligands with
identical affinities for a receptor, equation (8.22) reverts to the original
form of Clark’s equation (equation 8.19). This means that a normal response
curve is obtained and the drug acts as a full agonist. However, when
a
=
0 the percentage
response is zero and the drug is a full antagonist. Intermediate values between
1 and 0 for
a
indicate a partial agonist (Fig.).
In 1950s,
Stephenson discovered that a maximum response was obtained when only a
proportion of the available receptors were occupied. This discovery was in
direct conflict with Clark’s occupancy theory and led Stephenson to
independently propose a two-stage route for receptor action. Independently of
Ariens he proposed that the binding of a ligand to a receptor produced a
stimulus (S) that was related to tissue response. The magnitude of the stimulus
depends on both the affinity of the ligand for the receptor and its efficacy (e).
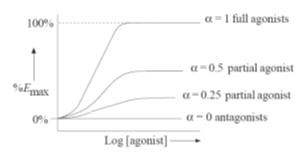
Figure
A pictorial representation of the variation of
dose–response curves with the value of
a.
Values for
a
between 1 and 0 correspond
to drugs that act as partial agonists, the degree of partial agonism depending
on the value of
a.

As a result,
Clark’s equation (8.19) was modified to:

Equation (8.23)
shows that ligands with an
e
value of zero
will have no biological response. Consequently, full antagonists will have an
e
value of zero. To obtain a positive response
e
must have a positive value and so agonists and partial agonists will
have positive
e
values. Moreover, the
higher the positive value, the greater the response (Fig.) and the lower the
dose of agonist [D] required to achieve the maximum response. This means that
agonists with a high efficacy will produce a maximum response even though they
do not occupy all of the available receptor sites. Unoccupied receptors are
known as
spare receptors. Their presence increases the sensitivity of a
receptor to other ligands. It is now known that cells can contain several
thousand receptors of a particular type. This number can increase (upregulation)
or decrease (downregulation). These changes
may be brought about by both pathological and physiological cell stimuli. They
can affect drug response. For example, an increase (upregulation) in the number
of receptors (RT)
moves the drug response curve to a lower concentration, whilst a
decrease will move it to a higher concentration (Fig.).
The effects on the dose–response curves of increasing
e
and
RT

Stephenson
observed that the magnitude of the response was not linearly related to the
stimulus. This lead to a further modification of Clark’s equation to:
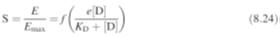
where
f
is a function known as the
transducer function.
The transducer function represents the properties of the signal transducer
mechanism that links the signal from the ligand to the tissue response and is a
characteristic of the responding tissue. As a result, the same ligand could have
different transducer functions when it is bound to different tissues. This
difference would explain why a ligand may act as an agonist in one tissue but as
a partial agonist in a different tissue even though it is acting on the same
receptor. Furthermore, differences in the transducer functions of different
ligands acting on the same receptor in the same tissue would also explain why
their relative potencies may be different. Potency depends on both the
ligand–receptor complex and its efficacy. It is used to compare the relative
effectiveness of different drugs and is defined as:

where
e
is the
intrinsic efficacy,
which is the efficacy per receptor, that is:

Since intrinsic
efficacy is independent of the total number of available receptors, a drug with
the same value for
e
in different tissues is likely to be
acting on the same receptor in those tissues. Conversely, if the values are
different, the drug is likely to be acting on different receptors in the
different tissues.
Drugs for Treating Endoparasitic and Ectoparasitic
Infestations
Adverse
hygienic conditions favor human infestation with multicellular organisms
(parasites). Skin and hair are colonization sites for arthropod ectoparasites,
such as insects (lice, fleas) and arachnids (mites). Against these, insecticidal
and arachnicidal agents, respectively, can be used. Endoparasites invade the
intestines or even internal organs and are mostly members of
the phyla
of flatworms and roundworms. They are combated with anthelmintics.
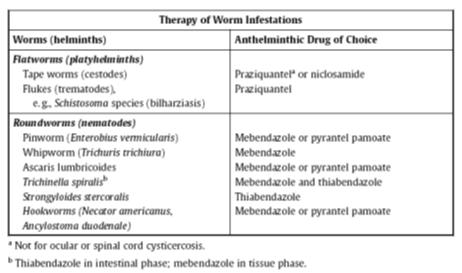
Antihelmintics.
As shown in the
table, the newer agents,
praziquantel
and
mebendazole,
are adequate for the treatment of diverse worm diseases. They are generally well
tolerated, as are the other agents listed.
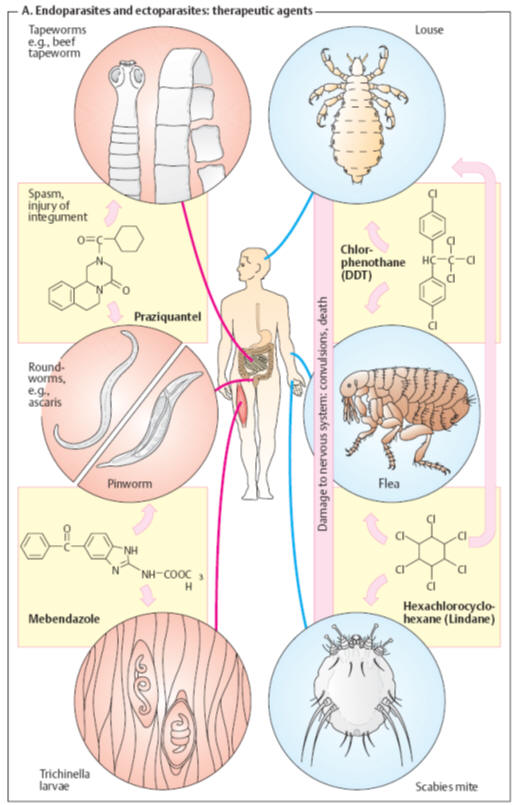
Insecticides.
Whereas fleas
can be effectively dealt with by disinfection of clothes and living quarters,
lice and mites require the topical application of insecticides to the infested
subject.
The
following agents act mainly by interfering with the activation or inactivation
of neural voltage-gated insect sodium channels.
Chlorphenothane
(DDT) kills
insects after absorption of a very low amount, e. g., via foot contact with
sprayed surfaces (contact insecticide). The cause of death is nervous system
damage and seizures. In humans DDT causes acute neurotoxicity only after
absorption of very large amounts. DDT is chemically stable and is degraded in
the environment and the body at extremely slow rates. As a highly lipophilic
substance, it accumulates in fat tissues. Widespread use of DDT in pest control
has led to its accumulation in food chains to alarming levels. For this reason,
its use has now been banned in many countries.
Lindane
is the active
γ-isomer
of hexachlorocyclohexane. It also exerts a neurotoxic action on insects (as well
as humans). Irritation of skin or mucous membranes may occur after topical use.
Lindane is active also against intradermal mites (Sarcoptes scabiei,
causative
agent of scabies), besides lice and fleas. Although it is more readily degraded
than DDT, it should be used only as a second line agent with appropriate
precautions. In the United Kingdom its use for head lice has been banned; in the
United States it is not recommended in young children and is contraindicated in
premature infants.
Permethrin, a synthetic
pyrethroid, exhibits similar antiectoparasitic activity and may be the drug of
choice owing to its slower cutaneous absorption, fast hydrolytic inactivation,
and rapid renal elimination.
Antifungal agents
Fungal infections
or
mycoses
may generally be divided
into either superficial or systemic mycoses. Superficial mycoses affect the
skin, nails, scalp and mucous membranes, while systemic mycoses affect internal
tissues and organs. Since the middle of the twentieth century there has been an
increase in both superficial and systemic mycoses. A significant degree of this
increase is believed to be due to medical treatments, such as the use of
antibiotics, radiotherapy, immunosuppressant drugs and steroids, which suppress
a patient’s immune system. It is believed that this supression allows the fungal
microorganisms to flourish. Fungal infections caused in this manner are referred
to as
opportunistic fungal infections.
Opportunistic infections also occur in conditions such as AIDS where the immune
system is suppressed by the disease.
Fungal
microorganisms are believed to damage the cell membrane, leading to a loss of
essential cellular components. This may result in inflammation of the infected
tissue, which in some cases may be severe. Antifungal agents counter myoses by
both
fungistatic
and
fungicidal
action. Fungistatic
action occurs when a drug prevents the fungi reproducing, with the result that
it dies out naturally, whilst fungicidal action kills the fungi. The suffixes -static
and -cide
are widely used to
indicate these general types of action.
Fungal
microorganisms differ from other microorganisms in that they consist of
eukaryotic cells with a chitin cell wall. This means that their chemical
structures and biochemistry are similar to those of humans. Consequently, it is
more difficult to design drugs that would selectively target these fungi.
However, there are some differences that can be utilised. For example, human
cell membranes contain cholesterol but those of fungi contain ergosterol. A
number of antifungal drugs are believed to act by blocking the biosynthesis of
ergosterol in fungi.
Azoles
The azoles are a
group of substituted imidazoles that exhibit fungistatic activity at nanomolar
concentration and fungicidal activity at higher micromolar concentrations
(Fig.). They are active against most fungi that infect the skin and mucous
membrane. Azoles are also active against some systemic fungal infections,
bacteria, protozoa and helminthic species.
(a)
Examples of the structures of some active 1,3-diazoles. Note the common
structural features. (b) Examples of azoles
based on 1,2,4-triazole ring systems
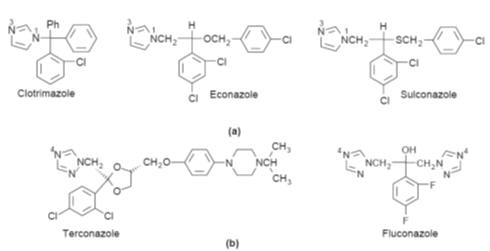
An outline of the biosynthesis of ergosterol
In common with
most drugs, the azoles are believed to act at a number of different sites, all
of which contribute to their fungicidal action. However, their main point of
action is believed to be the inhibition of some of the cytochrome P-450 oxidases
found in the membranes of the microorganisms. In particular, azoles have been
linked to inhibition of the enzyme 14a-sterol
demethylase (P-450DM), which is essential for the biosynthesis of ergosterol,
the main sterol found in the fungal cell membranes (Fig.). It is believed that
nitrogen at position 3 of the imidazole rings (Fig. a) and nitrogen at position
4 of the triazole rings (Fig. b) bind to the iron of the haem units found in the
enzyme, thereby blocking the action of the enzyme. This appears to lead to an
accumulation of 14a-methylated sterols such as
lanosterol in the membrane, which is thought to increase the membrane’s
permeability, allowing essential cellular contents to leak causing irreversible
cell damage and death. However, the precise details of the mode of action of
azoles have yet to be fully elucidated. Azoles also inhibit the P-450 oxidases
found in mammalian steroid biosynthesis, but in mammals much higher
concentrations than those necessary for inhibition of the fungal sterol 14a-demethylases are usually required.
Structure–action studies have shown that a weakly basic imidazole or
1,2,4-triazole rings substituted only at the N-1 position are essential for
activity. The substituent must be lipophilic in character and usually contains
one or more five- or six-membered ring systems, some of which may be attached by
an ether, secondary amine or thioether group to
the carbon chain.
The more potent compounds have two or three aromatic substituents, which in the
more potent compounds are singly or multi-chlorinated or -fluorinated at
positions 2, 4 and 6. These non-polar structures give the compounds a high
degree of lipophilicity, and hence membrane solubility.
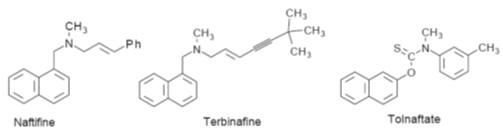
Allylamines and related compounds
Allylamines are
synthetic derivatives of 3-aminopropene (Fig. ) developed from naftifine. They
are weak bases, their hydrochlorides being only slightly soluble in water. The
allylamine group appears to be essential for activity.
Allylamines are believed to act by inhibiting squalene epoxidase, the
enzyme for the squalene epoxidation stage in the biosynthesis of ergosterol in
the fungal membrane (Fig.). This leads to an increase in squalene concentration
in the membrane with subsequent loss of membrane integrity, which allows loss of
cell contents to occur. Tolnaftate, although it is not an allyl amine, appears
to act in a similar fashion. However, allylamines do not appear to significantly
inhibit the mammalian cholesterol biosynthesis.
Examples of the structures of allylamines. Naftidine and terbinafine are used as
fungicides to treat deratophytes and filamentous fungi but only have a
fungistatic action against pathogenic yeasts
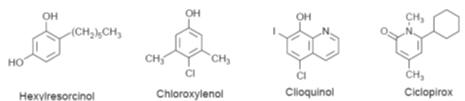
Phenols
There are
numerous phenolic antifungal agents (Fig.). They are believed to destroy
sections of the cell membrane, which results in the loss of the cellular
components and the death of the cell. The mechanism by which this destruction
occurs is not known. Ciclopirox is not a phenol but appears to have a similar
action. However, at low concentrations it has been shown to block the movement
of amino acids into susceptible fungal cells.
Examples of phenolic compounds used as antifungal agents
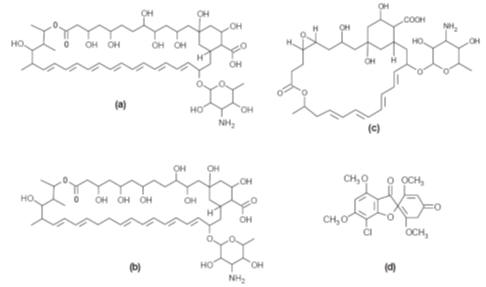
Antibacterial antifungal agents
A number of
antibiotics are important antifungal agents. They are mainly polyenes such as
amphopterin B, nystatin and natamycin (Fig.). However, the antibiotic
griseofulvin (Fig.d) is also active. The smaller polyenes (26-membered ring)
exhibit both fungistatic and fungicidal action at the same concentration. In
contrast, the larger ring polyenes (38-membered ring) show fungistatic action at
lower concentrations and fungicidal action at higher concentrations. This
indicates some differences in their mode of action. All the polyenes are
believed to act by binding to the cell membrane, causing leakage of the
cytoplasmic contents and cell lysis. It is thought that amphopterin B binds to
the ergosterol found in the cell membranes of microfungi to form a transmembrane
channel that allows the leakage of the cell contents. Unfortunately, it also
appears to act in the same way with the cholesterol found in human cell
membranes, which accounts for its toxic side effects in humans when administered
by parenteral routes. The poor water solubility of polyenes means that they are
difficult to administer by parenteral routes. However, amphopterin B may be
administered parentally using micelle formulations. Consequently, their
difficulty of administration and often unpleasant side effects results in
polyenes being used mainly in topical preparations.
Griseofulvin is a
fungistatic agent. It acts by preventing the infestation of new tissue as that
tissue is formed. This is a slow process and so its use is only successful if
the patient sticks rigidly to the prescribed drug regimen. Griseofulvin is used
to treat systemic infections and has few side effects. It has a poor water
solubility and so its oral adsorption and hence its effectiveness will depend on
how the dose is formulated.
(a)
Amphopterin B, first isolated from Streptomyces nodosus by Gold
et al. (b)
Nystatin, first isolated from Streptomyces noursei by Hazen and Brown. (c)
Natamycin, first isolated from Streptomyces natalensis by Struyk
et al.
(d)
Griseofulvin,first isolated from Penicillium griseofulvium by Oxford
et al.
Chemotherapy of Viral Infections
Viruses
essentially consist of genetic material (nucleic acids) and a capsular envelope
made up of proteins, often with a coat of a phospholipid (PL) bilayer with
embedded proteins. They lack a metabolic system and depend on the infected cell
for their growth and replication. Targeted therapeutic suppression of viral
replication requires selective inhibition of those metabolic processes that
specifically serve viral replication in infected cells.
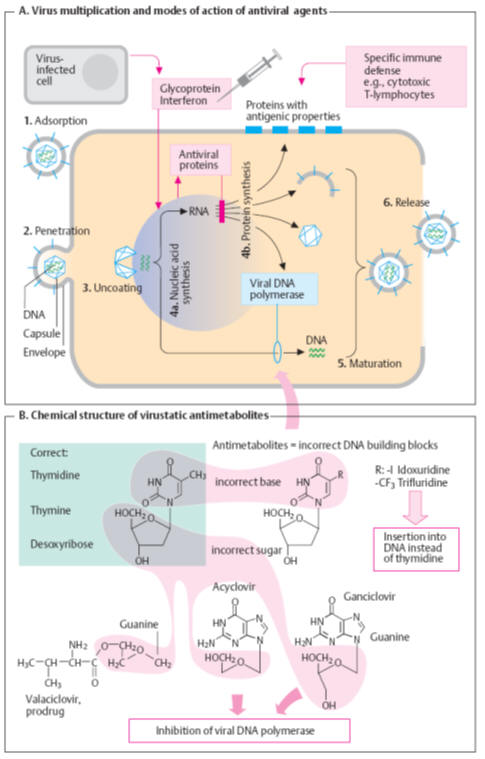
Viral replication as exemplified by
herpes simplex
viruses (A)
1. The
viral particle attaches to the host cell membrane (adsorption) via envelope
glycoproteins that make contact with specific structures of the cell membrane.
2. The
viral coat fuses with the plasmalemma of host cells and the nucleocapsid
(nucleic acid plus capsule) enters the cell interior (penetration).
3. The
capsule opens (“uncoating”) near the nuclear pores and viral DNA moves into the
cell nucleus. The genetic material of the virus can now direct the cell’s
metabolic system.
4a.
Nucleic acid synthesis: The
genetic material (DNA in this instance) is replicated and RNA is produced for
the purpose of
protein synthesis.
4b. The
proteins are used as “viral enzymes” catalyzing viral multiplication (e. g., DNA
polymerase and thymidine kinase), as capsomers, or as coat components, or are
incorporated into the host cell membrane.
5.
Individual components are assembled into new virus particles (maturation).
6.
Release of daughter viruses results in spread of virus inside and outside the
organism. With herpesviruses, replication entails host cell destruction and
development of disease symptoms.
Antiviral mechanisms (A)
The
organism can disrupt viral replication with the aid of cytotoxic T-lymphocytes
that recognize and destroy virus-producing cells (presenting viral proteins on
their surface) or by means of antibodies that bind to and inactivate
extracellular virus particles. Vaccinations are
designed
to activate
specific immune defenses.
Interferons
(IFN) are
glycoproteins that, among other products, are released from virus-infected
cells. In neighboring cells, interferon stimulates the production of “antiviral
proteins.” These inhibit the synthesis of viral proteins by (preferential)
destruction of viral DNA or by suppressing its translation. Interferons are not
directed against a specific virus, but have a broad spectrum of antiviral action
that is, however, species-specific. Thus, interferon for use in humans must be
obtained from cells of human origin, such as leukocytes (IFN-α),
fibroblasts (IFN-β), or
lymphocytes (IFN-γ). Interferons are used in the treatment of
certain viral diseases, as well as malignant neoplasias and autoimmune diseases;
e. g., IFN-α
for the
treatment of chronic hepatitis C and hairy cell leukemia; and IFN-β
in severe herpes virus infections and multiple sclerosis.
Virustatic antimetabolites
are “false” DNA building blocks (B)
or nucleosides. A nucleoside (e. g., thymidine) consists of a nucleobase (e. g.,
thymine) and the sugar deoxyribose. In antimetabolites, one of the components is
defective. In the body, the abnormal nucleosides undergo bioactivation by
attachment of three phosphate residues.
Idoxuridine
and congeners
are incorporated into DNA with deleterious results. This also applies to the
synthesis of human DNA. Therefore, idoxuridine and analogues are suitable only
for topical use (e. g., in herpes simplex keratitis).
Chemotherapy of Malignant Tumors
A tumor
(neoplasm) consists of cells that proliferate independently of the body’s
inherent “building plan.” A malignant tumor (cancer) is present when the tumor
tissue destructively invades healthy surrounding tissue or when dislodged tumor
cells form secondary tumors (metastases) in other organs. A cure requires the
elimination of all malignant cells (curative therapy). When this is not
possible, attempts can be made to slow tumor growth and thereby prolong the
patient’s life or improve quality of life (palliative therapy). Chemotherapy is
faced
with the
problem that the malignant cells are endogenous and almost lacking in specific
metabolic properties.
Cytostatics (A)
Cytostatiscs
are cytotoxic substances that particularly affect proliferating
or dividing (mitotic) cells. Rapidly dividing malignant cells are preferentially
injured. Damage to mitotic processes not only retards tumor growth but also may
initiate
apoptosis
(programmed cell death). Tissues with a low mitosis rate are largely unaffected;
likewise, most healthy tissues. This, however, also applies to malignant tumors
consisting of slowly dividing differentiated cells. Tissues that have a
physiologically high mitosis rate are bound to be affected by cytostatic
therapy. Thus,
typical adverse effects
occur.
Loss of hair
results from injury to hair follicles;
gastrointestinal disturbances, such as diarrhea, from inadequate
replacement of enterocytes whose lifespan is limited to a few days;
nausea and vomiting
from
stimulation of area postrema chemoreceptors; and
lowered resistance to infection
from weakening of the immune
system. In addition, cytostatics cause
bone marrow depression. Resupply of blood cells depends on the
mitotic activity of bone marrow stem and daughter cells. When myeloid
proliferation is arrested, the short-lived granulocytes are the first to be
affected (neutropenia), then blood platelets (thrombopenia) and, finally, the
more long-lived erythrocytes (anemia).
Infertility
is caused by suppression of spermatogenesis or follicle
maturation. Most cytostatics disrupt DNA metabolism. This entails the risk of a
potential genomic alteration in healthy cells (mutagenic
effect).
Conceivably, the latter accounts for the occurrence of leukemias several years
after cytostatic therapy (carcinogenic
effect). Furthermore, congenital malformations are to be expected when
cytostatics must be used during pregnancy (teratogenic
effect).
Cytostatics
possess different
mechanisms of action.
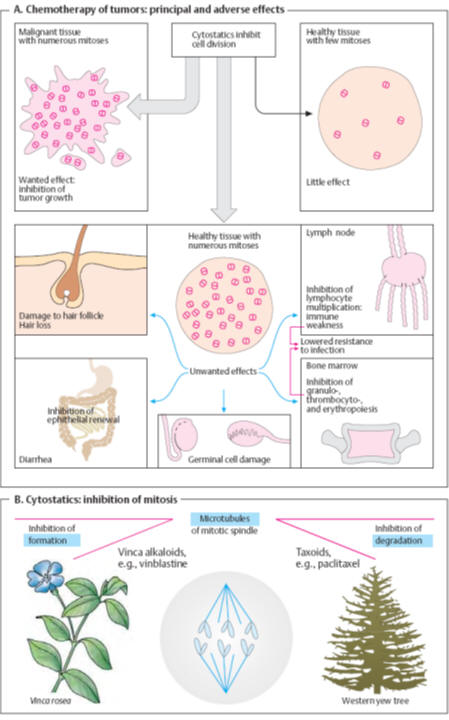
Damage to themitotic spindle (B)
The
contractile proteins of the spindle apparatus must draw apart the replicated
chromosomes before the cell can divide. This process is prevented by the
so-called
spindle poisons
that arrest mitosis at metaphase by disrupting the assembly into spindle threads
of microtubuli. These consist of the proteins
α-and
β-tubulin. Surplus tubuli are broken down,
enabling the tubulin subunits to be recycled. The vinca alkaloids,
vincristine
and
vinblastine
(from the
periwinkle plant, Vinca rosea),
inhibit the polymerization of tubulin subunits into microtubuli. Damage to the
nervous systemis a predicted adverse effect arising from injury to
microtubule-operated axonal transport mechanisms.
Paclitaxel, from the bark
of the pacific yew (Taxus brevifolia),
inhibits disassembly of microtubules and induces formation of atypical ones, and
thus impedes the reassemblage of
tubulins
into properly functioning microtubules.
Docetaxel
is a semisynthetic derivative.
Inhibition of DNA and RNA synthesis (A)
Mitosis
is preceded by replication of chromosomes (DNA synthesis) and increased protein
synthesis (RNA synthesis). Existing DNA (gray) serves as a template for the
synthesis of new (blue) DNA or RNA. De-novo synthesis may be inhibited by the
following mechanisms.
Damage to the template (1)
Alkylating cytostatics
Alkylating cytostatics
are reactive compounds that transfer alkyl
residues into a covalent bond with DNA. For instance,
mechlorethamine
(nitrogen mustard) is able to
cross-link double stranded DNA on giving off its chlorine atoms. Correct reading
of genetic information is thereby rendered impossible. Other alkylating agents
are
chlorambucil,
melphalan,
thio-TEPA,
cyclophosphamide,
ifosfamide,
lomustine,
and
busulfan.
Specific adverse reactions include irreversible pulmonary fibrosis due to
busulfan and hemorrhagic cystitis caused by the cyclophosphamide metabolite
acrolein (preventable by the uro-protectant mesna = sodium
2-mercaptoethanesulfonate).
The
platinum-containing compounds
cisplatin
and
carboplatin
release
platinum, which binds to DNA.
Cystostatic
antibiotics
insert themselves into the DNA double
strand; this may lead to strand breakage (e. g., with
bleomycin). The
anthracycline antibiotics daunorubicin
and
adriamycin
(doxorubicin)may induce
cardiomyopathy. Bleomycin can also cause pulmonary fibrosis.
Induction of strand breakage
may result from inhibition
of topoisomerase.
The epipodophyllotoxins
etoposide
and
tenoposide
interact with
topoisomerase II, which functions to split, transpose, and reseal DNA strands;
these agents cause strand breakage by inhibiting resealing. The “tecans”
topotecan
and
irinotecan
are derivatives
of camptothecin from the fruits of a Chinese tree (Camptotheca acuminata). They
inhibit topoisomerase I, which induces breaks in single-strand DNA.
Inhibition of nucleobase synthesis (2)
Tetrahydrofolic acid (THF) is required for the synthesis of both purine bases
and thymidine. Formation of THF from folic acid involves dihydrofolate reductase
(p. 274). The
folate analogues aminopterin
and
methotrexate
(amethopterin) inhibit enzyme activity.
Cellular
stores of THF are depleted. The effect of these antimetabolites can be reversed
by administration of folinic acid (5-formyl-THF, leucovorin, citrovorum factor).
Hydroxyurea
(hydroxycarbamide) inhibits
ribonucleotide reductase that normally converts ribonucleotides
into
deoxyribonucleotides subsequently used as DNA building blocks.
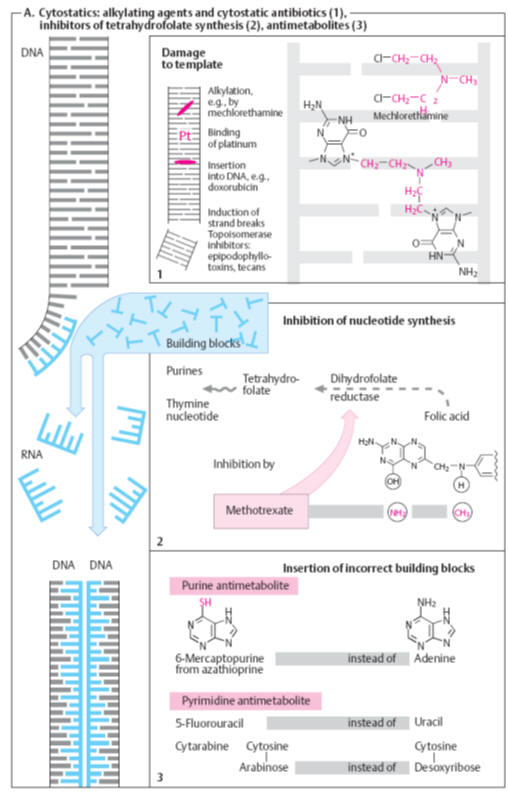
Incorporation of false building blocks (3)
Unnatural
nucleobases (6-mercaptopurine;
5-fluorouracil)
or nucleosides with incorrect sugars (cytarabine)
act as antimetabolites. They inhibit DNA/RNA synthesis or lead to synthesis of
missense nucleic acids. 6-Mercaptopurine results from biotransformation of the
inactive precursor
azathioprine.
The uricostatic
allopurinol
inhibits the degradation of 6-mercaptopurine such that coadministration of the
two drugs requires dose reduction of the latter.
Combination therapy
Cytostatics are frequently administered in complex therapeutic regimens designed
to improve efficacy and tolerability of treatment.
Supportive therapy
Cancer
chemotherapy can be supported by adjunctive medications. Thus, 5-HT3 serotonin
receptor antagonists (e. g., ondansetron) afford effective protection against
vomiting induced by highly emetogenic drugs such as cisplatin. Bone marrow
depression can be counteracted by granulocyte and granulocyte/macrophage
colony-stimulating factors (filgrastim and lenograstim and molgramostim,
respectively).
Immunomodulation
The control of disease by immunologic means has two objectives: the development
of immunity and the avoidance of undesired immune reactions. Immunostimulation
in a drug-induced immunosuppression model and immunosuppression in an
experimental hyperreactivity model by the same preparation can be said to be
true immunomodulation. Immunomodulators are biological response modifi ers
(BRMs), used to treat cancer, which exert their antitumor effects by improving
host defense mechanisms against the tumor. They have a direct antiproliferative
effect on tumor cells and also enhance the ability of the host to tolerate
damage by toxic chemicals that may be used to destroy the cancer. Modulation of
immunity was previously attempted with glucocorticoids and cytotoxic drugs such
as cyclophosphamide. It is now recognized that immunomodulatory therapy could
provide an alternative to conventional chemotherapy for a variety of diseased
conditions, especially when the host’s defense mechanisms have to be activated
under the conditions of impaired immune responsiveness or when a selective
immunosuppression has to be induced in situations such as infl ammatory
diseases, autoimmune disorders, and organ/bone marrow transplantation. All three
classes of immunomodulators—biologicals, chemical, and cytokines—will continue
to play a major role in advancing and improving the quality of treatment of
several human as well as animal diseases. There is need for further research to
better understand the biochemical mechanisms involved in immunoregulation to
maximize the benefi ts of chemical immunomodulators as single agents or
adjuvants in cancer therapy.
Ethnopharmacology and Botanical Immunomodulators
There are two major ways of bioprospecting natural products for investigation.
The first is the classical method, which relies on phytochemical factors,
serendipity, and random screening approaches. The second method uses traditional
knowledge and practices as the drug discovery engine. Known as the
ethnopharmacology approach,
this method is time and cost-effective and may lead to better success than
routine random screening. Various ethnopharmacological agents are under
investigation as immunomodulators. Traditional Chinese medicine, Japanese Kampo,
Indian Ayurveda, and such are becoming important bioprospecting tools. Ayurveda
gives a separate class of immunomodulating botanicals named
Rasayanas. Ayurveda, one
of the most ancient and yet living traditions practiced idely in India, Sri
Lanka, and other countries, has a sound philosophical and experiential basis.
India has about 45,000 plant species; medicinal properties have been assigned to
many to several thousands. Ayurveda has detailed descriptions of over 700 herbs
and 400,000-registered Ayurvedic practitioners routinely prescribe them,
particularly for treatment of chronic disease conditions. Considerable research
on pharmacognosy, chemistry, pharmacology, and clinical therapeutics has been
carried out and the Ayurvedic database has detailed descriptions of over 700
medicinal plants. Rasayanas are nontoxic herbal preparations or individual herbs
used to rejuvenate or attain the complete potential of a healthy or diseased
person in order to prevent diseases and degenerative changes that lead to
disease. Pharmacodynamic studies on Rasayana botanicals have suggested many
possible mechanisms, such as nonspecific and specific immunostimulation,
free-radical quenching, cellular detoxification, cell proliferation, and cell
repair. Ayurveda (with particular reference to botanicals) may play an important
role in modern health care, particularly where satisfactory treatment is not
available. There is a need to evaluate the potential of Ayurvedic remedies as
adjuvant to counteract side effects of modern therapy and compare the
cost-effectiveness of certain therapies vis-à-vis modern therapeutic schedules.
Adaptogens or Adjustive Medicine
Most of the synthetic chemotherapeutic agents available today are
immunosuppressants, are cytotoxic, and exert a variety of side effects. N. V.
Lazarev, who developed the concept of a state of nonspecifically increased
resistance of an organism (SNIR), laid down the theoretical basis for separation
of a new group of medicinal substances. The medicinal substances causing SNIR
were named
adaptogens. Generally, adaptogens are those drugs that enable one to
withstand the stress and strain of life, impart immunity to give protection
against diseases, postpone aging, and improve vigor, vitality, and longevity.
The concept is also referred to as
adjustive medicine. The concept of adjustive remedies has been
difficult to prove experimentally. A bifunctional information exchange network
between the nervous and immune systems is established by specific receptors for
humoral substances on cells of nervous and immune systems. In particular,
neuroregulators (neurotransmitters and neuromodulators) can modulate specific
immune system function(s), and immunoregulators (immunomodulators) can modulate
specific nervous system function(s). Acute and chronic inflammatory processes,
malignancy, and immunological reactions stimulate the synthesis and release of
immunomodulators in various cell systems.
Botanicals with Adaptogenic Activity
Mistletoe Lectin
Defined nontoxic doses of the galactoside-specific mistletoe lectin (mistletoe
lectin-I, a constituent of clinically approved plant extract) have
immunomodulatory potencies. The obvious ability of certain lectins to activate
nonspecific mechanisms supports the assumption that lectin-carbohydrate
interactions may induce clinically beneficial immunomodulation. Randomized
multicenter trials are being performed to evaluate the ability of complementary
mistletoe lectin-I treatment to reduce the rate of tumor recurrences and
metastases, to improve overall survival and the quality of life, and to exert
immunoprotection in cancer patients under tumor-destructive therapy.
Achyranthes Bidentata
Achyranthes bidentata
polysaccharide (ABP) root extract (25 to 100 mg/kg, day
_1 to 7) could inhibit tumor growth (S-180) by 31 to 40%. A
combination of cyclophosphamide and ABP increased the rate of tumor growth
inhibition by 58%. ABP could potentiate LAK cell activity and increase the Con
A–induced production of tumor necrosis factor (TNF-â) from murine spleenocytes. The S-180 cell membrane content of
sialic acid was increased, and phospholipid decreased after ABP had acted on
cells for 24 hours. Data suggest that the antitumor mechanism of ABP may be
related to potentiation of host immunosurveillance mechanism and the changes in
cell membrane features.
Chemoprotection and immunomodulation.
Chemoprotection
Modern cancer therapy produces substantial acute and chronic toxicity, which
impairs quality of life and limits the effectiveness of treatment. Recent
clinical and laboratory data suggest that repair of treatment-related injury is
a multiphase and continuous process providing multiple opportunities for
pharmacologic intervention. A host of agents (toxicity antagonists) are under
development that modulate normal tissue response or interfere with mechanisms of
toxicity. Although significant challenges remain, the routine application of
such agents promises to reduce treatment-related morbidity substantially and
potentially to allow treatment intensification in high-risk disease. The concept
of site-specific inactivation of cytotoxic anticancer agents has been explored
with numerous modalities. The goal of such chemoprotection is to improve the
therapeutic ratio of an agent by selectively reducing its toxicity in
non-tumor-bearing tissue, which is target for dose-limiting toxicity.
Furthermore, a chemoprotectant cannot add new toxicities that might otherwise
limit the administration of maximally tolerated doses of chemotherapeutic agent.
Drug Targets and Current Trends
Chemoprotection and cytoprotection are studied under preventive oncology and are
interchangeable terms in cancer chemotherapy. Preventive oncology applies
pharmacological agents to reverse, retard, or halt progression of neoplastic
cells to invasive malignancy. Cancer chemoprevention is one of the newer
approaches in the management of cancer. Epidemiological observations,
preclinical animal pharmacology, knockout models, cancer cell lines, and
clinical trials have shown the efficacy of this approach. Many drug targets are
under clinical development; prostaglandin pathway, estrogen receptor modulation,
gluthathione peroxidase inhibition, and immunomodulation appear promising.
Celecoxib, tamoxifen, retinoids, rexinoids, selenium, tocopherols, and
mofarotene are some of the promising leads and are in clinical development. New
opportunities in clinical chemoprevention research include investigating
chemopreventive effects of phytochemicals. Safer immunomodulating agents
suitable for long-term therapy remain an unmet therapeutic need. Figure gives a
schematic overview of cytoprotection and immunomodulation.
Botanical Immunomodulators as Chemoprotectants
Withania Somnifera
Withania somnifera
is an official drug mentioned in the
Indian Herbal Pharmacopoeia
and
Ayurvedic Pharmacopoeia.
Studies indicate that
W. somnifera
(ashwagandha) (WS) possesses anti-inflammatory, antitumor,
antistress, antioxidant, immunomodulatory, hemopoietic, and rejuvenating
properties. The chemistry of WS has been studied extensively, and over 35
chemical constituents have been identified, extracted, and isolated. The
biologically active chemical constituents are alkaloids (isopelletierine,
anaferine), steroidal lactones (withanolides, withaferins), saponins containing
an additional acyl group (sitoindoside VII and VIII), and withanolides with a
glucose at carbon 27(sitoindoside IX and X).
The suppressive effect of cyclophosphamide-induced toxicity by WS extracts was
observed in mice. Administration of WS extracts signifi cantly reduced
leucopenia induced by cyclophosphamide treatment, resulting in an increase in
bone marrow cellularity. Administration of
W. somnifera
extract for 5 days along with cyclophosphamide
(CTX) (1.5 mmol/kg body weight, i.p.) reduced the CTX-induced urotoxicity.31
Treatment of
W. somnifera
resulted in the enhancement of interferon gamma (IFN-gamma),
interleukin-2 (IL-2), and granulocyte macrophage colony stimulating factor
(GM-CSF), which were lowered by cyclophosphamide administration. Pharmacodyanmic
studies reveal that the major activity of
W. somnifera
may be due to
the enhancement of cytokine production and stem cell proliferation
and its differentiation. These studies indicate that
W. somnifera
could reduce the
cyclophosphamide toxicity and its usefulness in cancer chemotherapy.
The major activity of WS may be due to stimulation of stem cell proliferation,
indicating the fact that WS could reduce cyclophosphamide toxicity and its
usefulness in cancer chemotherapy. WS was also shown to prevent lipid
peroxidation (LPO) in stress-induced animals, indicating its adjuvant as well as
chemoprotectant activity. Glycowithanolides, consisting of equimolar
concentrations of sitoindosides VII to X and withaferin A, isolated from the
roots of WS were evaluated for protection in iron-induced hepatoxicity in rats.
Ten days of oral administration of these active principles, in graded doses (10,
20, and 50 mg/kg), resulted in attenuation of hepatic lipid peroxidation (LPO)
and the serum enzymes alanine aminotransferase, aspartate aminotransferase, and
lactate dehydrogenase during iron-induced hepatoxicity.58 Antistress activity
observed with
W.
somnifera
will be an additional benefit, along with
chemoprotectant activity.
Tinospora Cordifolia
Tinospora cordifolia
is
widely used in ayurvedic medicines and is known for its immunomodulatory,
antihepatotoxic, antistress, and antioxidant properties. It has been used in
combination with other plant products to prepare a number of ayurvedic
preparations. The chemistry has been studied extensively, and its chemical
constituents can be broadly divided into alkaloids, diterpenoids, steroids,
flavanoids, and lignans. Reviews have appeared on quaternary alkaloids and
biotherapeutic diterpene glucosides of
Tinospora
species. Much of the work has been carried out on berberine,
jatrorrhizine, tinosporaside, and columbin. Extracts of
T. cordifolia
(TC)
have been shown to inhibit lipid peroxidation and superoxide and hydroxyl
radicals in vitro. The extract was also found to reduce the toxic side effects
of cyclophosphamide (25 mg/kg, 10 days) in the mice hematological system by
free-radical formation as seen from total white cell count, bone marrow
cellularity, and esterase-positive cells. The active principles of TC were found
to possess anticomplementary and immunomodulatory activities. TC is reported for
its various immunopharmacological activities (e.g., inhibition of C3-convertase
of the classical complement pathway). Humoral and cell-mediated immunity were
reported for cardioside, cardifolioside A, and cardiol and their activation was
more pronounced with increasing incubation time.61 Extracts of
T. cordifolia
has
been shown to inhibit lipid peroxidation and superoxide and hydroxyl radicals in
vitro. The extract was also found to reduce the toxic side effects of
cyclophosphamide (25 mg/kg, 10 days) in the mice hematological system by
free-radical formation as seen from total white cell count, bone marrow
cellularity, and esterase-positive cells.
Botanical Immunomodulators as Antitumor Agents
Plant products have contributed several novel compounds that possess promising
antitumor activity. Crude extract of
W.
somnifera
root has a strong tumoricidal and tumor growth
inhibitory activity. Withaferin A, an alkaloid isolated from the leaves,
has been reported to show marked tumor inhibitory activity in vitro against
cells derived from human carcinoma of nasopharynx and experimental mouse tumors.
A single i.p. dose of withaferin A injection 24 or 48 hours after Ehrlich’s
ascites tumor transplantation produced an immediate growth reduction in 3 to 80%
of mice, followed by complete disappearance of tumor cells in the peritoneal
cavity of the surviving mice with no signs of tumor development. However,
withaferin A is toxic in mice, with an LD50 of 54 mg/kg body weight after an
i.p. injection. An effective dose of crude extract was much higher (a cumulative
dose of more than 10 g—750 mg/kg daily for 15 days) with less toxicity than
reported doses of purified withaferin A and withanolide D, which exhibited toxic
effects. Crude extract included a range of chemicals (e.g., a few flavanoids,
several alkaloids, and other withanolides) in addition to withaferin A. In the
ayurvedic system of treatment, dry powders or crude extract is used, and hence
the effects observed may not be attributed to a single component. The rationale
for this type of treatment is that the toxicity of an active component may be
counteracted by another component, which may not have the desired therapeutic
property. Resinous material from a methanol extract and orange-colored oil from
a petroleum ether extract of
Semecarpus anacardium
Linn. F. have been found to possess antitumor properties in a P388
lymphocytic leukemia model. Acetylated oil of
Semicarpus anacardium, which itself does not possess antitumor
activity against experimental transplantable tumors, enhances the antitumor
effect of anticancer drugs such as mitomycin-C, 6-mercaptopurine, and
methotrexate when used in combination against P388 and S180 (ascites) tumor
systems.
Extracts of
T. cordifolia
have been shown to inhibit lipid peroxidation and superoxide and
hydroxyl radicals in vitro. The extracts were also found to reduce the toxic
side effects of cyclophosphamide administration in mice. Moreover, their
administration partially reduced elevated lipid peroxides in serum and liver as
well as alkaline phosphatase and glutamine pyruvate transaminase thus indicating
the value of
Tinospora
extracts in reducing the chemotoxicity induced by free
radical–forming chemicals. Crude saponins obtained from shoots of
Asparagus racemosus
[asparagus crude saponins (ACSs,] were found to have antitumor activity. They
inhibited the growth of human leukemia HL-60 cells in culture and macromolecular
synthesis in a dose- and time-dependent manner. The ACS in the range 75 to 100
mg/mL was cytostatic; at concentrations greater than 200
mg/mL it was cytocidal to HL-60 cells. ACSs at 6 and 50
mg/mL inhibited the synthesis of DNA, RNA, and protein in HL-60
cells by 41, 5, and 4% or by 84, 68, and 59%, respectively. The inhibitory
effect of ACSs on DNA synthesis was irreversible.