
Drug Metabolism
A
major problem in all drug discovery programs is to discover compounds with good
pharmacokinetics. Although it is possible to examine the metabolism of the drug
in animals, it has often been difficult to predict what would happen in man. The
obvious implications of drug metabolism are an effect on half-life
in vivo
and the production of toxic metabolic products.
In seeking to establish an effective dose for a new drug, the clinician needs to
know what ranges of abilities humans will have to metabolize the drug and what
effect the drug will have on the metabolizing enzymes. Failure to metabolize the
drug may lead to overdose, whereas rapid metabolism could lead to lack of
clinical benefit. Equally, inhibition of the metabolism of another drug could
cause problems in a patient receiving several medications.
A
large proportion of the metabolizing enzymes are members of the P450
superfamily28 and a large number of these genes have now been cloned and their
metabolic potential determined. Increasingly, the enzymes are being expressed in
microbial systems, e.g. yeast, where their ability to metabolize the drug can be
evaluated. In a few years, it would not be surprising if all new drugs were
“typed” for their complete P450 metabolism profile.
Equally, their metabolic products can be identified and their biological
activity and toxicity determined. An additional application is likely to be the
P450 genotyping of patients. As “poor metabolizers” become recognized in the
population, the problem is often found to be mutations in one or more of their
P450 genes. Once identified, such mutations are easily screened for and it is
entirely likely that some degree of P450 “profiling” will take place for
patients in the future. Armed with knowledge on the metabolic fate of new drugs,
the physician will then be able to prescribe the best drug for an individual
depending on their P450 profile. This individualization of drug therapy based on
genetic information is known as pharmacogenomics. There is a massive effort
currently underway to identify and characterize polymorphisms in a wide variety
of genes, including drug receptors and effectors, in addition to drug
metabolizing enzymes. It is hoped that the correlation of these polymorphisms
with clinical outcomes and drug effects across a population will allow for the
prediction of the safety, efficacy and toxicity of both established drugs and
new drugs in development and, thereby, a reduction in the size and expense of
clinical trials.
Receptors
Molecular recognition refers to the affinity of biological
molecules to their receptor and is basis for biological processes. The physical
basis for association between drug and their receptors (affinity) is important
for the drug action, hence for drug design. The drug-receptor interactions
(molecular recognitions) are key for the biological responses. The molecular
interaction pattern (affinity) determines the molecular function as agonist and
antagonist.
The macromolecular protein entities (sensing elements)
interacting with the endogenous chemical messengers (e.g., acetylcholine,
histamine) as well as exogenous molecules (e.g., paracetamol) are known as
receptors. The term receptors include cellular macromolecules such as regulatory
proteins (enzymes) trans-membrane glycoproteins (ion channel),
mucopolysaccharides and nucleic acids. The tissue (functional elements) is
composed of the lipid layers sandwiched between protein. These proteins adopt
different conformations in bio-phase and acts as a receptor. The molecular
interaction between the endogenous compounds and receptor (target) coordinates
the cellular functions. The receptors with no defined pharmacological activity
is known as orphan receptor.
Receptor Types
The receptor mediated cellular communication may produce rapid
(synaptic transmission), intermediate time scale (adrenergic action) and
prolonged (thyroid action) effects. The functional variation is mainly dependent
on the receptor molecular structure. Based on these mechanisms, receptors are
grouped into four
sub-families.
1. G-protein coupled receptors (GPCRs)
2. Ion channels
3. Transmembrane catalytic receptors
4. Nuclear receptors
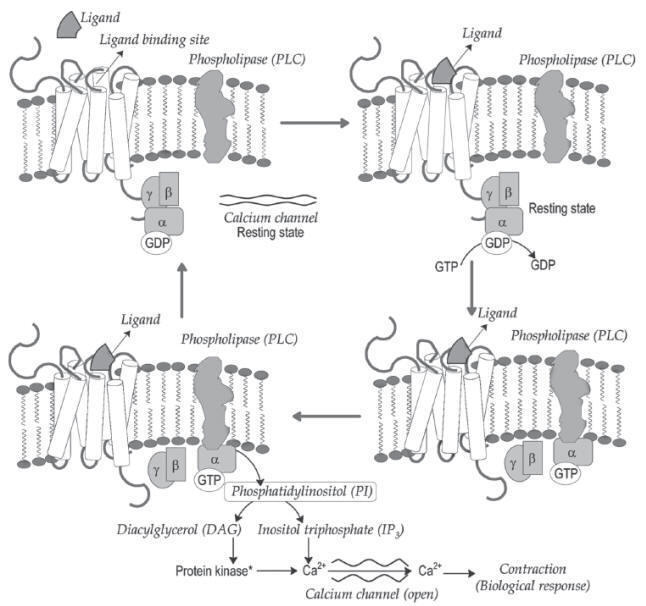
G-protein Coupled Receptors (GPCRs)
G-protein coupled receptors (GPCR)
are large membrane-bound proteins, which are known as
metabotropic/7-transmembrane (7-TM) receptors. These receptors transduce their
bio-chemical signal through activating guanine nucleotide binding proteins
(G-protein). G-proteins are constituted by á, â and ã subunits (hetero
trimeric). The subunits are anchored to the receptor through prenylation
reaction. The subunit of trimer has binding affinity for guanine nucleotides
(GTP and GDP). In resting state (before bonding to receptor), it has affinity
for GDP and hold in it.
GPCR
mediated adenylyl cyclase (AC) activation and resulting biological contraction.
The receptor extracellular region has amino terminus and loops.
This region is called as ligand binding site. The carboxylic end of receptor
exists intracellularly, which has binding sites for G-proteins. The ligand
binding activates the receptor (induces conformational changes). The binding of
ligand to the extracellular ligand binding site facilitates the recognition of
G-protein binding site by G-protein. G-protein (trimer) binds to the receptor in
between the sixth and seventh trans-membrane region (serine and threonine rich
region).
The conformational changes on trimer upon binding to the receptor
triggers the dissociation of GDP molecule from á-subunit and replacement of GTP
(GDPGTP exchange). The GTP binding to the trimer (abg) favours the
dissociation of trimer into monomeric unit bound with GTP and the dimer (bg). The
released
aGTP
monomer binds to the particular target enzyme and produces
activation or inhibition of the target function. The
a-subunit has
GTP hydrolysing potential and converts
a-subunit bound GTP into GDP
through dephosphorylation reaction. The
a-subunit bound with GDP molecules
reunion with
bg dimer. The cycle repeats and elicits the biological response
(Fig.).
GPCRq
mediated phospholipase-C (PLC) activation and resulting biological contraction.
These receptors are
grouped into four families.
1. GPCRs:
The
aGTP
binding to the enzyme (target) facilitates the second messenger
synthesis. The activated adenylyl cyclase (AC) generates cyclic adenosine
monophosphate (cAMP) from adenosine triphosphate (ATP) and activated guanylyl
cyclase (GC) generates cyclic guanosine monophosphate (cGTP) from guanosine
triphosphate (GTP). The resulting cAMP and cGMP activates the intracellular
protein kinase and favours phosphorylation mediated cellular function.
2. GPCRi:
The
aGTP binding
to the enzyme (target) inhibits the functions of AC and GC. This binding
stabilizes the conformation of the enzyme and no catalysis for the generation of
cAMP and cGMP.
3. GPCRq:
The
aGTP binding
activates the enzyme phospholipase-C (PLC). The activated enzyme catalyzes the
generation of second messengers phosphatidylinositol (PI2) and diacylglycerol
(DAG) from inositol triphosphate (IP3) (Fig.).
4. GPCR12
This receptor
is expressed in renal convoluted tubules
(GPR12):and
facilitates the Na+/H+ exchange function.
Ion Channels
Ion channels
undergo conformational switch with reference to the physical and chemical
changes (electro chemical gradient) in their environment. The conformational
changes decides the transport of ions through the channels (Fig.). The different
types of ion channels present in the bio-phase are as follows
1. Ligand
(transmitter)-Gated Ion Channels (LGICs)
2.
Second-messenger-Gated Ion Channels (SGICs)
3.
Voltage-Gated Ion Channels (VGICs)
Structural depiction for the ion transport through ion channels.

1.
Ligand-Gated Ion Channels (LGICs):
It contains
four membrane-spanning domains. These channels are also known as
ionotropic/metabotropic receptor. The neurotransmitters namely, gamma-amino
butyric acid (GABA), glutamate, N-methyl-D-aspartic acid (NMDA),
alpha-amino-3-hydroxy-5-methyl isoxazole-4propionic acid (AMPA), glycine and
serotonin are the substrates for the LGICs. The binding of selective ligands to
the ion channels favours the opening of ion channel and influx of particular
ions.
2.
Second-messenger-Gated Ion Channels (SGICs):
The second
messengers namely cAMP, cGMP, IP3 and DAG influences the opening and closing of
calcium (Ca2+) and potassium (K+) channels.
3.
Voltage-Gated Ion Channels (VGICs):
The influx of
sodium (Na+), calcium (Ca2+) and efflux
of potassium (K+) ions are
responsible for the generation of depolarization and action potential. The
electro chemical gradient of intra and extracellular regions facilitates the
movement of selective ions through the ion channels. The channels permits the
movement of ions. The brief depolarization introduced by the glutamate receptors
favours the opening of NMDA receptor and produce long depolarization.
Drugs:
Local
anaesthetics, anti-convulsants and calcium channel blockers.
Changes in membrane (chemical) potential regulates the opening of
the channels. Voltage gated ion channels open, when cell membrane is
depolarized. Depolarization caused by the opening of Na+
influx initiates the opening Ca2+ channels. This kind of channel opening is
short-lasting even if the depolarization is maintained. It produces influx of Ca2+
and initiates oxidative phosphorylation.
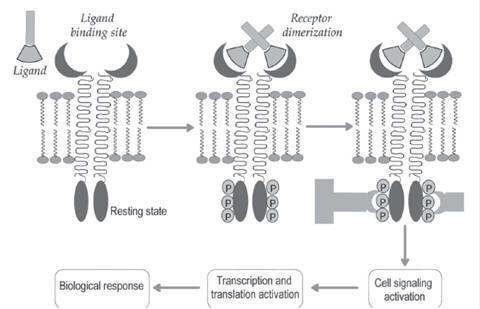
Transmembrane Catalytic Receptors
/ Enzyme-Coupled Receptors
These
receptors contain three domains namely, extracellular ligand binding domain, a
single membrane-spanning domain and intracellular effector domain with enzymatic
activity. Peptide hormones (insulin, leptin), epidermal growth factors,
platelet-derived growth factors and arterial natriuretic factors activate the
functions of their receptors. The ligand binding to the receptor promotes the
receptor dimerization (association of cellular effector domain) and permits the
auto phosphorylation of receptor tyrosine residues. The phosphorylated tyrosine
residues are binding sites for intracellular proteins (adapter proteins).
(a) The
activated catalytic receptors results in cellular functions such as opening of
ion channels and gene expression.
(b) In case
of enzyme-coupled receptors, signal transducers and activators of transcription
(STATs) bonds to the phosphorylated receptors. The enzyme Janus Kinase (JAK)
phosphoralytes the STAT and translocates into nucleus (Fig.). This initiates the
gene transcription, which is essential for the cell growth, cell division and
cell differentiation.
Biochemical pathway for transmembrane catalytic receptor.
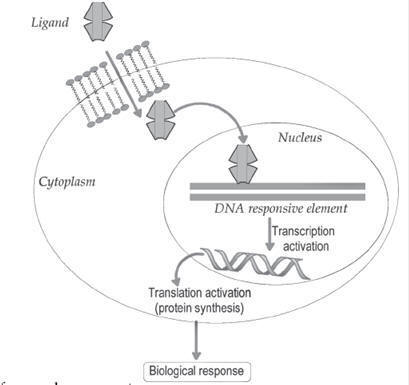
Nuclear
Receptors
Nuclear receptors are composed of a three functional domains
namely modulator protein binding domain (heat-shock protein-90, HSP-90), DNA
binding domain and ligand binding domain. The thyroid receptor (TR), Vitamin D
receptor (VDR), retinoic acid receptor (RXR) and peroxisome
proliferator-activated receptor (PPAR) belongs to this nuclear receptor
category. The natural ligands include nitric oxide, steroid hormones, thyroid
hormones, retinoic acid and vitamin D. The lipophilic nuclear receptor ligands
readily cross the cell membrane. The binding of ligands to the nuclear receptor
favours the dissociation of heat-shock protein-90 (HSP-90). This ligand bound
receptor diffuses into the nucleus and binds with DNA response element. This
promotes the gene transcription (RNA polymerase function) and biological
functions (Fig.).
Biochemical pathway for nuclear receptor.
Receptor Theories
The difference in the biological potential difference of
molecules can be explained through the different drug-receptor theories
(hypothesis). These theories explain about the pattern of drug-receptor binding.
Paul Ehrlich proposed side-chain theory for the molecular (dye) interactions.
The theory states that all cells have side chains for binding with molecules
(with specific functional groups). The nutrient-capturing structures (side
chains) of cells bind to molecule (endogenous and exogenous) through specific
groups and elicit characteristic biological response. The cell extends more side
chains after the initial binding with fewer molecules. This statement is valid
for the release of antibodies (side chains) with respect to the antigens. These
side chains are later identified as amino acids and are named as receptors. This
hypothesis is primer for drug-receptor theories.
The different drug receptor theories are listed below and
explained in the following section.
1. Occupancy theory
2. Rate theory
3. Induced fit theory
4. Macromolecular-perturbation theory
5. Activation-aggregation theory
Occupancy Theory
The occupancy theory states that the receptor mediated biological
response is directly proportional to the number of receptors occupied. A
relationship between drug concentration and the proportion of receptors occupied
was explained through occupancy theory. The earlier hypothesis of Clark and
Gaddum were basis for the evolution of occupancy theory.
Clark (1933), proposed that only a
small population of the receptor (affected tissue) could be occupied by the
drug.
Gaddum (1937) explained the
binding of two mutually exclusive drugs with opposite functions at the same
receptor. This is known as drug antagonism theory.
A tissue response is dependent on
the fraction of specific receptors occupied by the drug. In many instances a
very small portion of tissues (receptors) are occupied by the drug. The maximum
response results when more receptors are occupied by a drug.
Human
organism is made up of
3 × 1023
molecules.
1 mole (molecular weight = 100 Dalton) of drug contains 6.023 ×
1023
molecules. 1 mg of drug (molecular weight = 100 Dalton) contains
![]()
Therefore each drug molecule would act on 106
molecules of the receptor (3 × 1023 / 3 × 10–19).
The law of mass action describes
the receptor occupation. The interaction between a drug (D) and a receptor (R)
is responsible for the drug effect (E), which can be expressed as
![]()
ka
= rate constant for association; kd= rate constant for dissociation;
D = drug; R =
receptor;
DR =
drug-receptor complex; E = biological effect
The dissociation of drug from the
complex terminates the therapeutic effect.
The structural features of the
molecules decide their molecular affinity towards the receptor and initiates
pharmacological function.
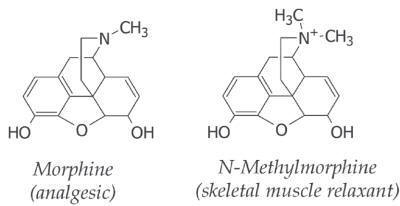
Structurally specific drugs:
The
structural difference (even minor) among the analogues alters their biological
effects (activity and potency). These drugs are called as structurally specific
drugs. The natural alkaloid morphine has analgesic
potential. But the N-methylmorphine produces skeletal muscle
relaxant effect. This reflects the influence of chemical structure in their
receptor specificity.
Structurally non-specific drugs:
The
physicochemical properties (not structural) of drugs are also determinant factor
for their pharmacological function. These drugs are called as structurally
non-specific drugs. Gaseous general anesthetics (e.g., halohthane and
isoflurane) and antacids (e.g., aluminium hydroxide and calcium carbonate) are
examples. The halothane and isoflurane accumulate (not by binding with receptor)
in the membranes and elicit their general anesthetic function. The antacids
produce their effect by acid neutralization function.
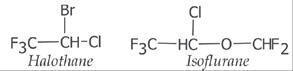
Schild ‘Dose Ratio’ Concept
The number of drug-receptor complexes responsible for the
observed biological response is concentration dependent.
Ariens-Stephenson Concept:
The occupancy theory provides qualitative description of
drug-receptor interaction and lacks quantitative descriptions. The independent
observation of Ariens and Stephenson explains that the ability of the
molecules to induce conformational change in the receptor is
responsible for the activity.
• Ariens (1954) introduced the term intrinsic activity to explain
about the strength of drug response.
• Stephenson (1956) introduced to the term efficacy to explain
about the strength of drug affinity with receptor.
Agonist and antagonist molecules possess complementary structural
features (desirable affinity) for the receptor. The agonist molecules upon
binding with receptor produce stimulant action, whereas the antagonist produces
no biological response. The agonist molecules have capacity to induce the
conformational change in the receptor upon binding, whereas antagonists do not
induce the conformational change. The binding affinity difference among the
agonists is reason for their biological potential variation.
Chaerniere Theory:
Rocha Silva
(1969) stated that the agonists and antagonists establish reversible binding on
the receptor. The theory proposes that pharmacological receptor has two sites
for binding with drugs.
(a) Specific site (critical):
The receptor sites which interacts with pharmacophore group.
(b) Non-specific site (non-critical):
The receptor sites which interact
to the non-polar groups of antagonists.
The antagonist competes with agonist and the interactions are
stronger (through hydrophobic, van der Waals and charge transfer interactions).
According to the hypothesis, the agonist (even in high concentrations) cannot
displace the antagonist due to their stronger interactions.
Rate Theory
Paton (1961) suggested that the rate of association between the
drug and receptor as an important factor in the drug response. The rate of
association and dissociation of drug-receptor complex rather than number of
receptors occupied is responsible for the observed pharmacological response. The
therapeutic effects of molecules are very much influenced by their rate of
association and dissociation. The activation of receptor is proportional to the
rate of drug-receptor association in unit time. The drug-receptor complex
strength has very less influence on its biological effect.
(a)
Agonists: In case of agonists, the rate of
dissociation is relatively faster than the rate of association (high
dissociation rate).
(b)
Antagonists: In case of
antagonists, the rate of association is faster than the rate of dissociation
(low dissociation rate). The drug dissociation from the drug-receptor complex
can be estimated through dissociation constant.
![]()
KD = dissociation constant; Df = concentration of free
drug; Rf
= concentration of free receptor; DR = drug-receptor complex;
Induced Fit Theory
The active binding site of the receptor is more flexible and can
be altered by the interactions of ligands. The binding of ligand to polymeric
protein induces conformational changes in one of the subunit. In several cases,
the binding of one molecular unit to the receptor binding pocket facilitates the
binding of another group by altering the receptor shape.
del Castillo and Katz (1957)
suggested that the drug exhibits its activity through two step mechanism. In the
first step, the drug generates intermediate drug-receptor conformation and in
the next step, it converts into active conformation. This active conformation
determines the drug response.
A receptor will undergo
conformational change as the ligand approaches it. In the induced conformation,
receptors will expose catalytic groups for binding to drug. Thus, the receptor
will initiate the biological response and returns to its original conformation
once the drug is released.
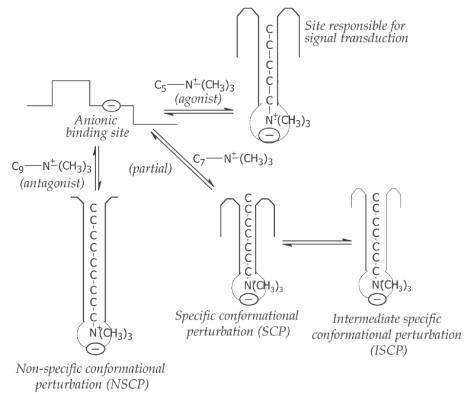
Macromolecular Perturbation Theory
Belleau (1964) described about the conformational adoptability of
the muscarinic (cholinergic) receptor. This hypothesis is known as
macromolecular perturbation theory. The molecular interaction between the drug
and macromolecule produce specific conformational perturbation (SCP) and
non-specific conformational perturbation (NSCP) in receptors.
1.
Specific conformational perturbation (SCP): The molecular perturbation occurs through the van der Waals
interaction. The agonists with intrinsic activity favor the SCP.
2.
Non-specific conformational perturbation (NSCP):
It involves hydrophobic interactions. The molecules with no
intrinsic activity (antagonists) generate NSCP. A partial agonist will induce
both SCP and NSCP.
Activation-Aggregation Theory
Stephenson (1956) proposed that the drug-receptor interaction can
be viewed as graded activation and the two-state model.
(a)
Graded activation:
Agonist molecules induce different
conformations in their receptor; hence produce different degree (strength) of
biological function.
Receptor conformational perturbation : specific conformational perturbation
(SCP) induced by agonist and non-specific conformational perturbation (NSCP)
induced by antagonist.
Activation-aggregation theory: Illustration for graded activation.

(b)
Two-state model:
The receptor exhibits two-state
namely active conformation (R*) and resting state conformation (R). The
receptors exhibit dynamic equilibrium between active and inactive form even in
resting state. The agonist molecules bind to active form of the receptor and
produce biological response. But the antagonist binds to inactive form of the
receptor and antagonizes (blocks) biological function. The partial agonists
preferentially bind to active form but also bind to inactive form; hence,
produces mixed response (less biological response).
Activation-aggregation theory: Illustration for two-state model.

Receptor Promiscuity
Molecules (agonists, inverse agonists, antagonists) exhibit
entirely different biological response in different bio-phase, even though the
binding receptor is same. This phenomenon is called as receptor promiscuity and
can be exemplified by the action of H1 and H2
blockers on histamine receptors in different locations of the
biological system.
Receptor Topology
The distances between two consecutive turns of á-helix of
proteins is 5.38 Å. The adjacent two peptide bonds are separated by the distance
of 3.61 Å. The chemical groups of several drugs keep one or another of these
distances or multiple of them. 1. In case of local anesthetics (procaine),
adrenergic blockers (piperoxan), cholinergic agents (acetylcholine),
spasmolytics (adiphine) and antihistamines (chlorpheniramine) the bond distance
between X and N atoms are 5.5 Å. 2. The distance between acetyl choline nitrogen
atom and carbonyl group is 5.16 Å. In cholinergic (carbachol) and
anticholinergic (ipratropium bromide) drugs the distance between carbonyl group
and nitrogen group is 7.2 Å (multiple of 3.61 Å).
Three-Dimensional Arrangement
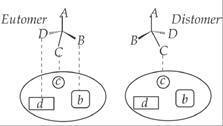
In the chiral environment, the
enantiomers of a drug behave differently and display different chemical and
pharmacologic behavior. The R-isomer of the drug will not bind in the similar
pattern, compared with the corresponding S-isomer. In the illustration given
below, the groups (B, C and D) present in active enantiomer (eutomer) interact
with their respective binding sites (b, c and d) and produces beneficial effect (Fig. 2.12). The difference in
three dimensional (3D) arrangement of groups present in the inactive enantiomer
(distomer) offers no binding or weak binding and results in no therapeutic
effect.
Illustration for the three-dimensional (3D) attachment for drug to the receptor.
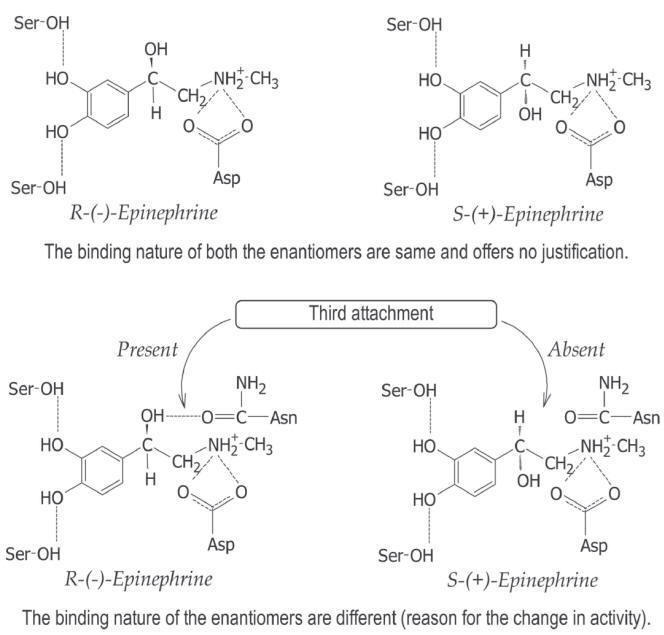
Three
point attachment:
In asymmetric molecule of R-(-)
epinephrine, the quaternary nitrogen, aromatic group and alcoholic hydroxyl
groups (â-hydroxyl group) are involved in the receptor binding. The
p-and
m-hydroxyl
groups (catechol group) determines the intensity of attachment (binding). The
alcoholic hydroxyl group of S-(+) epinephrine is in opposite orientation,
compared with R-(-) epinephrine (Fig. 2.13). The correct orientation of
alcoholic hydroxyl is important for the adrenergic activity.
Three
point attachment of epinephrine to the receptor.
Xenobiotics
Xenobiotics are chemicals or compounds that are
foreign to a biologic system. Exposure to xenobiotics may occur via the air,
water, diet, bedding, caging, and/or equipment, or may be pharmacologic agents
intentionally introduced as part of the routine conditioning or experimental
procedure. The effect or toxicity of a xenobiotic is based on the dose and
disposition. Absorption, distribution, biotransformation, and excretion all
affect xenobiotic disposition. In addition, host barriers, i.e., the skin,
lungs, and alimentary tract, and the physical and chemical composition of the
xenobiotic also affect its toxicity. The xenobiotic or its metabolites may cause
physiologic alterations in the animal and thus affect the outcome of the
experiment by altering immune function, and by acting as a mutagen and/or a
teratogen. Examples include aflatoxins; phytoestrogens; endocrine disruptors;
heavy metals such as lead, mercury, and cadmium; organochlorine insecticides;
and commonly administered anesthetic agents.
Inter-animal or inter-colony response variability to
xenobiotics may be attributable to differences in the intestinal microbiome of
individual animals or colonies as microbiotia may affect chemical metabolism by
altering biotransforming enzymatic activity, enterohepatic circulation,
absorption, direct chemical activation, the bioavailability of antioxidants and
environmental chemicals from feed, as well as gut motility, epigenetic
mechanisms, as well as genetic polymorphisms, gender and age.
Substances and sources of xenobiotics
|
Classification |
|||
|
Xenobiotic Substances |
Xenobiotic Sources |
||
|
characteristics |
classification |
example |
Direct sources: |
|
nature |
Natural |
Bacteriotoxins, zootoxins, phytotoxins, serotonin |
|
|
Synthetic |
Man-made substances, pesticides |
||
|
uses |
Active |
Pesticides, dyes, paints |
|
|
Passive |
Additives, carrier molecules |
||
|
physical state |
Gaseous |
Benzene, aerosol form |
|
|
Dust-form |
Asbestos powder |
||
|
Liquid |
Chemicals dissolved in water |
||
|
pathophysiological effects |
Tissue/organs |
Kidney toxins |
|
|
Biochemical |
Methemoglobin producing toxins |
||
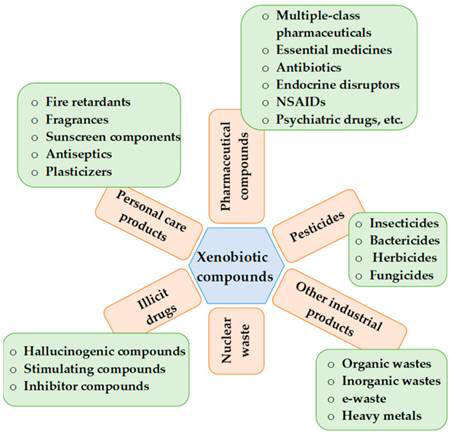
Xenobiotics include plant components, pharmaceutical drugs, pesticides,
cosmetic products, added food flavors, fragrances, etc. At higher concentrations
in environmental matrices, naturally occurring substances (endobiotics) may also
be considered as xenobiotics. Xenobiotics are categorized as pesticides,
pharmaceutical chemicals, personal care products, illicit narcotic
drugs/substances, industrial/commercial goods, and nuclear waste and can be
present in various environmental matrices. Xenobiotics are used by people and
directly or indirectly penetrate in the different environmental matrices
generating various metabolites and secondary products (some are even more
toxic). Finally, plants, algae, and aquatic organisms take up xenobiotics
leading to bioaccumulation, further causing biomagnification. One of the main
obstacles to the sustainable water availability in urban systems is the presence
of xenobiotics in aquatic ecosystems. In addition to the greater diversity of
enzymes present in complex and varied community of microflora, the chemical
distinctions between human and microbial transformations of ingested chemicals
result from different selection processes that cause these activities. While
host metabolism aids in the body’s elimination of xenobiotics, microbial changes
to these substances and their human metabolites frequently promote microbial
development by supplying nutrients or producing energy.
The amount of xenobiotics found in environmental matrices can be varied
from ng/L to g/L. In both humans and animals, long-term chronic exposure to even
little doses of xenobiotics may cause toxic, mutagenic, carcinogenic, or
teratogenic effects. These compounds may block the enzyme’s active site or
affect it in an allosteric way. Some xenobiotics including chlordecone,
dichlorodiphenyltrichloroethane (DDT), and dichlorodiphenyldichloroethylene
(DDE) show tendency to bioaccumulate, and even their low-level chronic exposures
can potentially have an adverse impact on cell signaling pathways. In order to
create safer molecules for use in human environment, knowledge of enhanced
molecular designs may be useful along with mechanism of xenobiotics’ action.
Biotransformation
Biotransformation
is the metabolic conversion of endogenous and xenobiotic chemicals to more
water-soluble compounds. Generally, the physical properties of a xenobiotic are
changed from those favoring absorption (lipophilicity) to those favoring
excretion in urine or feces (hydrophilicity). An exception to this general rule
is the elimination of volatile compounds by exhalation.
Chemical modification of a xenobiotic by biotransformation may alter its
biological effects. Some drugs undergo biotransformation to active metabolites
that exert their pharmacodynamic or toxic effect. In most cases, however,
biotransformation terminates the pharmacologic effects of a drug and lessens the
toxicity of xenobiotics. Enzymes catalyzing biotransformation reactions often
determine the intensity and duration of action of drugs and play a key role in
chemical toxicity and chemical tumorigenesis.
The metabolism of xenobiotics involves two sequential steps known as phase I and
phase II reactions.
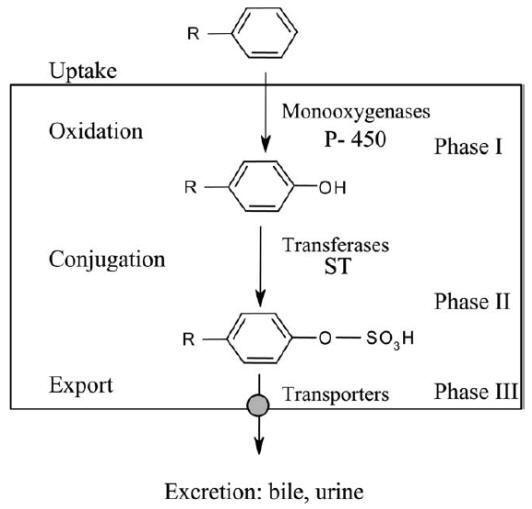
Sequential steps of drug biotransformation. After uptake by the cell,
a phenyl ring of a xenobiotic undergoes first a functionalization reaction
(oxidation, phase I). The hydroxyl metabolite is then conjugated by addition of
a sulfate group (phase II), before being exported from the cell by transporters
(phase III) and excreted. P-450, cytochromes P-450; ST, sulfotransferases.
During phase I, a
functionalization reaction
of the xenobiotic is achieved. New polar groups such as CO2H, OH or
NH2 are introduced or unveiled from pre-existing functions through oxidative,
reductive or hydrolytic reactions. The polar group created serves then as an
anchor point for the second metabolic step. The phase II reactions, known as
conjugation reactions, link
an endogenous, generally hydrophilic moiety, either to the original drug (if
polar functions are already present) or to the phase I metabolite. Common
endogenous groups are glucuronic acid, various amino acids, the tripeptide
glutathione, or sulfate. The water soluble conjugate is finally eliminated from
the cell by transport proteins (organic anion transporters, multidrug resistance
associated proteins), and finally excreted via the renal or the bile route. This
transport step is considered as phase III of drug metabolism.
The global result of phase I and II transformations should normally be the
inactivation and detoxication of the xenobiotic. However, innumerable examples
exist of metabolic activation. Phase I metabolite will possess its own activity
which will be similar, higher or different from that of the parent drug. Phase 2
metabolites are generally less subject to activation. Metabolic precursors can
even be designed to intentionally release the active species only
in vivo
upon transformation. Such compounds are called
prodrugs. Other metabolites, such as electrophiles, may
be highly reactive entities able to bind covalently to circulating or
intracellular proteins (formation of adducts), to enzymes (mechanism-based,
irreversible inactivation), or to DNA (mutagenic and carcinogenic compounds).
In this context, it is important to predict, at an early stage of a drug’s
development, the metabolic pathway, the type of metabolites formed and their
potential side/toxic effects. This challenge requires better knowledge, at a
molecular level, of the enzymes that are implicated in drug biotransformation
and the elucidation of the reaction mechanisms.
Drug biotransformation is catalysed by large families of enzymes (also known as
phase I and II drug metabolizing enzymes). These proteins are also implicated in
the metabolism of endogenous compounds. This situation, in which the drug or the
xenobiotic and the natural substrate compete toward the same protein, may lead
to cellular dysfunction and toxicity. Structural study of drug metabolizing
enzymes is an increasing area of research. With the development of sophisticated
technologies (genetic engineering: cloning, expression of cDNAs encoding these
proteins, site-directed mutagenesis), protein chemistry, molecular modelling,
X-ray crystallography, NMR etc., the organization of the active site can be
elucidated and the amino acids that play a crucial role for catalysis and in the
substrate recognition are identified. This chapter will illustrate the reaction
mechanisms that have been established and which account for the
biotransformation of drugs. Such information may help in predicting drug
metabolism and provide a rational basis for the design of safer drugs.
Phase I reactions
Oxidations. Most oxidative processes take place in liver microsomes and are
catalysed by mono-oxygenase enzymes known as mixed-function oxidases. These
processes require reduced nicotinamide-adenine dinucleotide phosphate, molecular
oxygen and a complex of enzymes in the endoplasmatic reticulum. The terminal
oxidizing enzyme is cytochrome P450, a hemoprotein. The notation ‘P450’ refers
to the ability of the reduced (ferrous) form of the hemoprotein to react with
carbon monoxide, yielding a complex with absorption peak at 450 nm. For each
molecule of substrate oxidized, one molecule oxygen is consumed; one oxygen-atom
is introduced into the substrate, and the other is reduced to form water. The
P450s represent a superfamily of enzymes. Initially it was believed that there
were only two forms, termed P450 and P448. Nowadays more than 30 different P450s
have been identified in humans. To unify the nomenclature, P450s are grouped in
families, designated by an arabic number, within which the amino acid sequence
homology is higher than 40%. The majority of P450s involved in drug metabolism
belong to three distinct families, CYP1, CYP2 and CYP3. Each P450 family is
further divided into subfamilies, designated by capital letters, which in
mammals contain proteins that share more than 55% amino acid sequence homology.
In each subfamily, specific enzyme are denoted by an Arabic number. Each
isoenzyme has more or less distinct substrate specificity requirements. Only six
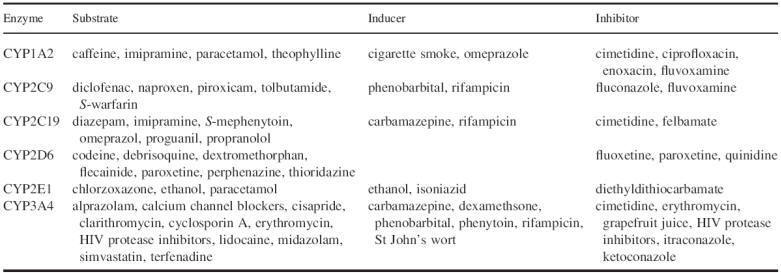
of the numerous cytochromes P450 play a major role in the metabolism of drugs in
common clinical use. Prominent among them in regard to the number of substrate
drugs are CYP3A4 and CYP2D6, with smaller numbers of drugs metabolized by
CYP2C9, CYP2C19, CYP1A2 and CYP2E1. Some selected substrates are listed in
Table. Cytochrome CYP1A2 is particularly involved in the metabolism of
environmental chemicals but also of drugs. CYP3A4 accounts for 30% of total P450
enzyme in the liver and is clinically the most important isoenzyme present in
the liver. It is also substantially expressed in the mucosal epithelium of the
intestine. Nearly 50% of all clinically used medications are metabolized by
CYP3A4. This explains to a large extentwhy this enzyme is involved in many
important drug interactions. Sometimes a single substrate is metabolized by a
single P450 enzyme, while other substrates can beoxidized to varying degrees by
multiple P450 enzymes.
Phase I reactions

In addition to cytochrome P450s, hepatic microsomes contain another class of
mono-oxygenases, the flavin containing mono-oxygenases (FMO). These enzymes
catalyse oxidation at nucleophilic nitrogen, sulfur and phosphorus atoms rather
than oxidation at carbon atoms, e.g. for phenothiazines, ephedrine, norcocaine
and the mono-ether and carbamate-containing pesticides. Some oxidations are
mediated by hepatic enzymes localized outside the microsomal system. Alcohol
dehydrogenase and aldehyde dehydrogenase, which catalyse a variety of alcohols
and aldehydes such as ethanol and acetaldehyde, are found in the soluble
fraction of the liver. Xanthine
oxidase, a cytosolic enzyme mainly found in the liver and in small intestine,
but also present in kidneys, spleen and heart, oxidizes mercaptopurine to
6-thiouric acid. Monoamine oxidase, a mitochondrial enzyme found in liver,
kidney, intestine and nervous tissue, oxidatively deaminates several naturally
occurring amines (catecholamines, serotonin), as well as a number of drugs.
Nonexhaustive list of substrates for, and inducers and inhibitors of
some human liver cytochrome P450 isoenzymes
Reduction. Reduction, for example azo- and nitro-reduction, is a less common
pathway of drug metabolism. Reductase activity is found in the microsomal
fraction and in the cytosol of the hepatocyte. Anaerobic intestinal bacteria in
the lower gastrointestinal tract are also rich in these reductive enzymes. A
historical example concerns ProntosilR, a sulfonamide prodrug. It is metabolized
by azo-reduction to form the active metabolite, sulfanilamide. Sulfasalazine is
also cleaved by azoreduction by intestinal bacteria to form aminosalicylate, the
active component, and sulfapyridine. Chloramphenicol is metabolized by
nitro-reduction to an amine in bacteria and in a number of tissues.
Hydrolysis. Hydrolysis of esters and amides is a common pathway of drug
metabolism. The liver microsomes contain non-specific esterases, as do other
tissues and plasma. Hydrolysis of an ester results in the formation of an
alcohol and an acid; hydrolysis of an amide results in the formation of an amine
and an acid. The ester procaine, a local anaesthetic, is rapidly hydrolysed by
plasma cholinesterases and, to a lesser extent, by hepatic microsomal esterase.
An example of an amide which is hydrolysed, is the antiarrhythmic drug
procainamide. Enalapril, a prodrug, is hydrolysed by esterases to the active
metabolite enalaprilate, which inhibits the angiotensin-converting enzyme.
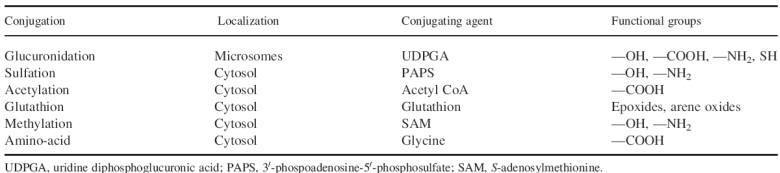
Phase II reactions
Compounds having polar constituents such as — OH, —NH2 or —COOH, or acquiring
them by a phase I reaction, may undergo a phase II or conjugation reaction. The
major conjugation reactions are listed in Table.
The reactive group of the drug interacts with endogenous compounds such
as glucuronic acid, sulfate, glycine, acetate or glutathione. Glucuronidation
and sulfation are the most common conjugation process. Knowledge of the
function, biochemistry, and molecular biology of the responsible enzymes, namely
the UDP glycosyltransferases (UGTs) and sulfotransferases (STs), has increased
extensively in recent
years. UGTs are membrane-bound enzymes and are located in endoplasmatic
reticulum, while STs are present in the cytosol. Both UGTs and STs comprise a
superfamily of enzymes; in humans at least eight UGTs and two STs have been
identified.
Conjugation reactions may involve an active, high energy form of the conjugating
agent, such as uridine diphosphoglucuronic acid (UDPGA) and acetyl CoA, which in
the presence of the appropriate transferase enzyme combines with the drug to
form the conjugate. For other conjugating reactions, the drug is activated to a
high-energy compound that then reacts with the conjugating agent in the presence
of a transferase enzyme. Glutathione, for example, reacts via the enzyme
glutathione-S-transferase
with reactive electrophilic oxygen intermediates of certain drugs, such as
paracetamol. Conjugates are usually pharmacologically inactive; they are more
hydrophilic than the parent compounds and are easily excreted by the kidneys or
the bile. Some conjugates, e.g. morphine-6-glucuronide and acetyl procainamide,
are pharmacologically active.
Stereoselective metabolism
Synthesis of a drug with an asymmetrical or chiral centre usually results in two
enantiomers, mirror images that cannot be superimposed. Such a 50:50 mixture is
called a racemate. The enantiomers of a racemic drug often differ in their
pharmacodynamic and/or pharmacokinetic properties as a consequence of
stereoselective interaction with optically active biological macromolecules.
Stereoselective metabolism of chiral xenobiotics is well recognized. Both phase
I and phase II metabolic reactions are capable of discriminating between
enantiomers. Stereoselective metabolism of chiral drugs implies the preferential
enzymatic removal of one enantiomeric form over the other. Stereoselective drug
metabolism may be divided in three groups: substrate stereoselectivity, product
stereoselectivity and substrate-product stereoselectivity.
1. Substrate stereoselectivity is characterized by the preferential enzymatic
metabolism of one enantiomer; metabolism can occur with retention or with loss
of stereoisomerism. Most examples of stereoselective metabolism belong to this
group. Several chiral nonsteroidal anti-inflammatory agents of the 2-aryl
propionic acid group undergo an unusual metabolic reaction whereby
R-enantiomers are inverted to the active
Santipodes. The extent of inversion varies
considerably depending on the drug, but is also species-dependent.
2. Product stereoselectivity is observed when a prochiral drug is preferentially
metabolized to one or more chiral products. There are only a few examples of
this type of stereoselectivity, e.g. the 5-hydroxylation of phenytoin and the
4-hydroxylation of debrisoquine, both with preferential formation of the
S-enantiomer of the hydroxylated product.
3. Substrate-product stereoselectivity: the enantiomers of a drug which possess
both asymmetrical and prochiral characteristics can undergo stereoselective
metabolism, whereby a second chiral centre is introduced. Examples are the
hydroxylation of perhexiline and the ketoreduction of warfarin.
Phase II reactions or conjugation reactions
Stereoselective metabolism is the most important process responsible for the
stereoselectivity observed in pharmacokinetics. Verapamil has received
considerable attention as an example of substrate stereoselective
pharmacokinetics in humans. After oral administration, the drug undergoes an
important stereoselective first pass metabolism, so that (-)-verapamil,
the active enantiomer, has a two to three times lower bioavailability than its
antipode. The (-)/(+) plasma concentration ratio is therefore higher after intravenous
than after oral administration.
Phase 3 Transporters
Once
xenobiotics have been converted into low-toxicity and water-soluble metabolites
by the combination of Phase 1 and Phase 2 reactions, these metabolites must be
transported against a concentration gradient out of the cell into the
interstitial space between cells, and then into the bloodstream for filtration
by the kidneys. The biggest hurdle is the transport of these hydrophilic
metabolites out of the cell. Charged Phase 2 metabolites will be effectively
“ion-trapped” in the cell, as the cell membrane is highly lipophilic and is an
effective barrier to the exit, as well as entry, of most hydrophilic molecules.
In addition, failure to remove the hydrophilic products of conjugation reactions
can lead to toxicity. Consequently, an array of multipurpose membrane-bound
transport carrier systems has evolved which can actively remove hydrophilic
metabolites and many other low-molecular-weight drugs and toxins from cells.
Thus, the term of Phase 3 metabolism has been applied to the study of this
essential arm of the detoxification process. Efflux transporters transport
hydrophilic substrates out of cells into interstitial fluid, blood, kidneys, and
the GIT. Influx transporters transport hydrophilic substrates into cells from
the bloodstream.
Membrane
transport plays an important role in the pharmacokinetic (administration,
distribution, metabolism and excretion) profiles of drugs. Membrane transporter
interaction leads to changes in transporter function and have effects on the
other co-administered drugs. Membrane transporters are proteins that govern the
mechanism by which drugs get into and out of cells in order to reach system
circulation as well as their sites of metabolism, storage and action.
Membrane transporters are found especially in epithelial tissue of the
liver, intestines and kidneys as well as the blood-brain barrier, testes and the
placenta.
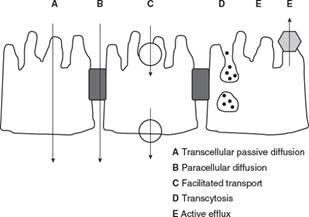
Mechanism of Membrane transport
There are five
different mechanisms by which drugs are transported across membranes.
Passive diffusion
Depends solely
upon a gradient concentration across the membrane and the lipophilic character
of both drug (unionized form) and the membranes.
Facilitated transport can be facultative or active.
Facultative
transporters: Membrane proteins that move drug molecules down a concentration
gradient without the use of a separate energy source.
Active
transporters: Membrane proteins that require an energy source (usually ATP) to
move drug molecules down and against a concentration gradient.
Paracellular transport
Drug molecules cross membranes by passing through the space between cells.
Transcytosis
The transport of extracellular drug molecules by their inclusion into cell
surface vesicles for transport across the interior of a cell.
Efflux transporters
Move drug molecules out of cells usually by an active mechanism.
ATP Binding Cassette (ABC) Transporters and inhibitors

Solute Carrier (SLC) Transporters and inhibitors

Cytochromes P450
Cytochromes P450 (CYP) are by far the most important xenobiotic- and
endobiotic-metabolizing monooxygenases. They represent up to 25% of the total
microsomal proteins and more than 50 cytochromes P450 monooxygenases are
expressed by humans. They are
involved in three main processes:
(1) drug metabolism;
(2) steroid metabolism (production of steroid hormones;
(3) haem degradation (conversion of haem into biliverdin and bilirubin).
Cytochromes P450 contain a molecule of haem, protoporphyrin IX, and a variable
protein of MW
,50 kDa. Cytochromes P450 form a very large group
of haemoproteins encoded by the CYP gene superfamily and classified in families
and subfamilies. The major xenobiotic metabolizing cytochromes P450 in humans
are found in family 1 (CYP1A1 and CYP1A2), family 2 (CYP2B6, CYP2C8, CYP2C9,
CYP2C18, CYP2D6 and CYP2E1), subfamily 3 (CYP3A), and family 4 (CYP4A9, CYP4A11
and CYP4B1). CYP3A4 is the most abundant and the most clinically important
cytochrome in humans, as it metabolizes up to 50% of the available drugs.
Cytochromes P450 belong to the haem-thiolate proteins in which the haem iron
fifth ligand is a thiolate group, generally a cysteine residue. Such protein
exhibits a Soret absorption band at 450 nm in the CO-difference spectrum of a
dithionite-reduced form.
The mechanism of the cytochrome P450 redox system is represented in Fig. In
microsomes, the two electrons necessary for monooxygenation are transferred by
NADPH cytochrome P450 reductase, the second electron in some cases coming from
NADH-cytochrome b5 reductase and cytochrome b5. In mitochondria, which also
contain cytochromes P450 devoted to the formation of steroid hormones from the
hydroxylation of cholesterol or to the biosynthesis of bile acids and vitamin D,
the electrons are supplied by the electron transfer chain composed of ferredoxin
(adrenoxin) and ferredoxin reductase (adrenoxine reductase).
In the resting state, the central iron atom of protoporphyrin IX is in a
hexacoordinated, ferric form. The substrate R — H binds reversibly to the enzyme
and the complex undergoes a reduction to the ferrous state. This allows
molecular oxygen to bind as a third partner. Following the second reduction
step, molecular oxygen is ultimately reduced to a hydroperoxide which is cleaved
with liberation of H2O and formation of a monooxygen known as oxene. The oxene,
which is electrophilic and quite reactive, can act on the substrate in a manner
which depends on the reactivity of the substrate itself. Thus, the oxene can
either:
(1) be transferred directly to the substrate (oxygen insertion or addition);
(2) remove an electron, or more frequently;
(3) pull a hydrogen radical away from the substrate and transfer back a formal
HO8
radical (a reaction known as oxygen rebound).
The latter is the mechanism by which RR0R00C — H is oxidized to RR0R00C — OH.
After release of the product, the regenerated cytochrome P450 is ready for a new
cycle. As illustrated below, the substrates to be oxidized are structurally
unrelated, the oxidation involving C, Si, N, P, S, Se and other atoms (Fig.).
The most common reaction catalysed by P450 is hydroxylation. However, it is also
involved in a wide spectrum of reactions including epoxidation,
O-,
N-, and
S-dealkylation,
deamination, desulfuration, dehalogenation and peroxidation.
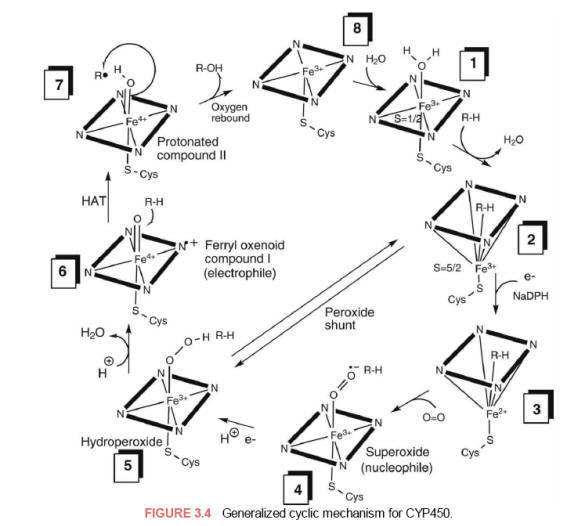
Step 1. The
resting [Fe3+-P450] complex binds reversibly with a molecule of the substrate
(RH) displacing the distal water resulting in a complex resembling
enzyme-substrate complex [Fe3+-P450*RH]. The binding of the substrate
triggers/facilitates the first one-electron reduction step from NADPH.
Step 2. The
substrate complex of [Fe3+-P450*RH] undergoes reduction to a [Fe2+-P450*RH]
substrate complex by an electron originating from its redox partner by the
flavoprotein [NADPHP450 reductase FNMH2/FADH complex].
Step 3. The
reduced [Fe2+-P450*RH] substrate complex readily binds dioxygen as the sixth
ligand of Fe2+ to form a [dioxy-Fe2+-P450*RH] substrate complex.
Step 4. The
[dioxy-Fe2+-P450*RH] complex rearranges by resonance because of the strong
electronegativity of O2 to form the [Fe3+-P450*RH-superoxide anion] complex.
Step 5. The
[Fe3+-P450*RH-superoxide] complex undergoes further reduction by accepting a
second electron from NADPH-P450 reductase to form the equivalent of a
two-electron reduced [peroxy-Fe3+-P450*RH] (hydroperoxide anion) complex (the
step where O2 is split into an oxygen atom). If the electron is not delivered
rapidly, this complex dissociates and is aborted (uncoupled) from subsequent
substrate hydroxylation at this step by xenobiotics which can cause release of
superoxide, which decomposes to hydrogen peroxide and dioxygen with regeneration
of the starting point of the cycle, the Fe3+-P450 complex.
Step 6. The
unstable [Fe3+-peroxy-P450*RH) complex undergoes heterolytic cleavage of
peroxide anion upon protonation to form water and a highly electrophilic
porphyrin-radical cation intermediate (ferryl oxenoid species, Fe4+=O) (Compound
I, the more favorable oxygen-cysteineporphyrin radical cation
resonance-stabilized complex). The Fe4+=O species represents the catalytically
active oxygenation species. One role of the cysteine sulfur ligand is thought to
be electron donation (push) that weakens the peroxide O-O bond, causing peroxide
bond scission to produce a highly reactive and strong oxidizing intermediate
(Compound I).
Step 7.
Abstraction of a hydrogen atom (HAT) from the substrate (RH) by the
electrophilic
[Fe4+=O*RH]
species to produce either a carbon-centered radical, radical addition to a
π-bond, or single electron transfer (SET) from a heteroatom to form a
heteroatom-centered radical-cation ferryl intermediate. Subsequent radical
recombination with the ferric-bound hydroxyl radical [Fe4+ *OH] (step 7/8,
oxygen rebound) or single electron-transfer (SET, deprotonation) yields the
carbon oxidized hydroxylated product (Compound II) and the regeneration of the
initial Fe3+-P450 enzyme complex.
The simplified cytochrome P450 redox cycle
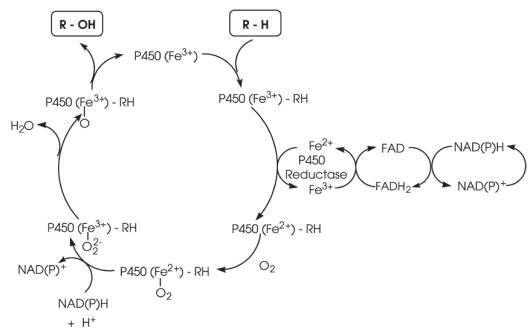
Major reactions of oxygenation catalysed by cytochrome P450.

Glutathione-S-Transferase (GST)
Glutathione (GSH,
g-glutamyl-cysteinyl-glycine)
is a thiol-containing tripeptide of capital significance in the detoxication and
toxication of drugs and other xenobiotics. Glutathione reacts with endogenous
and exogenous compounds in a variety of ways. First, the nucleophilic properties
of the thiol group make it an effective conjugating agent. Second, glutathione
can act as a reducing or oxidizing agent depending on its redox state (i.e. GSH
or GSSG). Furthermore, the reactions of glutathione can be enzymatic (e.g.
conjugations catalysed by glutathione-S-transferases, and peroxide reductions catalysed
by glutathione peroxidases) or non-enzymatic (e.g. some conjugation and various
redox reactions).
The glutathione transferase (GST) comprises multifunctional proteins coded by a
multigene family. These enzymes are both cytosolic and microsomal and function
as homodimers and heterodimers. They exist as four classes in mammals. The human
enzymes comprise the following dimers: A1-1, A1-2, A2-2, A3-3 (alpha class),
M1a-1a, M1a-1b, M1b-1b, M1a-2, M2-2, M3-3 (mu class), P1-1 (pi class), T1-1
(theta class), and three microsomal enzymes (MIC). The GST A1-1 and A1-2 are
also known as ligandin when they act as binding or carrier proteins, a property
also displayed by M1a-1a, M1b-1b and also by GSH (pi class).
GST are powerful detoxifying enzymes. The overexpression of GST that has been
demonstrated in some human cancer cells, such as breast cancer cells, is
associated with the multidrug resistance that impairs the efficacy of anticancer
drugs. In this regard, GST inhibitors or substrate competitors, such as the
diuretic drug ethacrynic acid, have been proposed as an adjuvant for cancer
chemotherapy, thus enhancing the cytotoxicity of alkylating drugs in cancer cell
lines. The nucleophilic character of glutathione is due to its thiol or rather
thiolate group. In deed if the thiol group of GSH (pKa
,
9) is largely protonated at physiological pH, the binding to the
enzyme is associated with the loss of the proton and to the electrophilic
stabilization of the thiolate group. A hydroxyl group of a serine (GST theta
class) or of a thyrosine residue (GST alpha, mu, pi classes) acts as hydrogen
donor to the sulfur of GSH whereby lowering the pKa of the thiol, leading to the presence of a predominantly ionized
form at physiological pH.
As a result, GSTs transfer glutathione to a very large variety of electrophilic
groups (R — X, see Fig.) in nucleophilic reactions categorized as either
substitutions or additions. With compounds of sufficient reactivity, these
reactions can also occur nonenzymatically. Once formed, glutathione conjugates
(GS-R) are seldom excreted as such, but usually undergo further
biotransformation. Cleavage of the glutamyl moiety by glutamyl transpeptidase
and of the cysteinyl moiety by cysteinylglycine dipeptidase or aminopeptidase M
leaves a cysteine conjugate (Cys-S-R) which is further
N-acetylated by cysteine-S-conjugate
N-acetyltransferase
to yield an
N-acetylcysteine
conjugate (CysAc-S-R). The latter type of conjugates are known as mercapturic
acids. Thesemay be either excreted or further transformed, since cysteine
conjugates can be substrates of cysteine-Sconjugate
b-lyase
to yield thiols (R-SH). These in turn can rearrange or be
S-methylated and then
S-oxygenated to yield thiomethyl conjugates (R-S-Me), sulfoxides
(R-SO-Me) and sulfones (R-SO2-Me).

Factors Affecting Metabolism
Drug therapy
is becoming oriented more toward controlling metabolic, genetic, and
environmental illnesses rather than short term therapy associated with
infectious diseases. In most cases, drug therapy lasts for months or even years,
and the problems of drug-drug interactions and chronic toxicity from long-term
drug therapy have become more serious. Therefore, a greater knowledge of drug
metabolism is essential. Several factors influencing xenobiotic metabolism
include:
Genetic polymorphism.
Genetic polymorphisms of drug metabolizing enzymes give rise to
distinct subgroups in the population that differ in their ability to perform
certain drug biotransformation reactions. Polymorphisms are generated by
mutations in the genes for these enzymes, which cause decreased, increased, or
absent enzyme expression or activity by multiple molecular mechanisms.
Individual differences in drug effectiveness (drug sensitivity or drug
resistance), drug-drug interactions, and drug toxicity can depend on racial and
ethnic characteristics impacting the population frequencies of the many
polymorphic genes and the expression of the metabolizing enzymes.
Pharmacogenetics focuses primarily on genetic polymorphisms (mutations)
responsible for interindividual differences in drug metabolism and disposition.
Genotype-phenotype correlation studies have validated that inherited mutations
result in two or more distinct phenotypes causing very different responses
following drug administration. The genes encoding for CYP2A6, CYP2C9, CYP2C19,
and CYP2D6 are functionally polymorphic; therefore, at least 30% of
P450-dependent metabolism is
performed by polymorphic enzymes.
For example, mutations in the CYP2D6 gene result in poor (PM),
intermediate (IM), or ultrarapid (UM) metabolizers of more than 30
cardiovascular and central nervous system (CNS) drugs. Thus, each of these
phenotypic subgroups experiences different responses to drugs extensively
metabolized by the CYP2D6 pathway ranging from severe toxicity to complete lack
of efficacy.
Physiologic factors.
Age
is a factor, as both very young and old have impaired metabolism. Hormones
(including those induced by stress), sex differences, pregnancy, changes in
intestinal microflora, diseases (especially those involving the liver), and
nutritional status can also influence drug and xenobiotic metabolism. Because
the liver is the principal site for xenobiotic and drug metabolism, liver
disease can modify the pharmacokinetics of drugs hepatically metabolized. Liver
disease affects the elimination half-life of some drugs but not of others, even
if they all undergo hepatic biotransformation. Some studies have shown that the
capacity for drug metabolism is impaired in chronic liver disease, which could
lead to unintentional drug overdosage. Because of the unpredictability of drug
effects in the presence of liver disorders, drug therapy in these circumstances
is complex, and more than usual caution is needed. Protein deficiency leads to
reduced hepatic microsomal protein and lipid metabolism, and oxidative
metabolism is decreased due to an alteration in endoplasmic reticulum (ER)
membrane permeability affecting electron transfer. Protein deficiency would
increase the toxicity of drugs and xenobiotics by reducing their oxidative P450
metabolism and clearance from the body.
Pharmacodynamic factors.
Dose, frequency, and route of administration, plus tissue distribution and
protein binding of a drug, affect its metabolism.
Environmental factors.
Competition of ingested environmental substances for metabolizing enzymes and
poisoning of enzymes by toxic chemicals such as carbon monoxide can alter drug
and other xenobiotic metabolism. Induction of enzyme expression (in which the
number of enzyme molecules is increased, while the activity is constant) by
other drugs and xenobiotics is another consideration. Environmental factors can
change not only the kinetics of an enzyme reaction but also the whole pattern of
metabolism, thereby altering bioavailability, pharmacokinetics, pharmacological
activity, and/or toxicity of a xenobiotic. Species differences in response to
xenobiotics must be considered in the extrapolation of pharmacological and
toxicological data from animal experiments to predict effects in humans.
Enzymes as drug targets
Several families of drugs do not act on receptors and their therapeutic
properties are attributed to inhibition or activation of enzyme activities. A
number of drugs in clinical use exert their effect by inhibiting a specific
enzyme present either in tissues of an individual under treatment or in those of
an invading organism. The basis of using enzyme inhibitors as drugs is that
inhibition of a suitable selected target enzyme leads to a build-up in
concentration of substrate and a corresponding decrease in concentration of the
metabolite, which leads to a useful clinical response. The type of inhibitor
selected for a particular target enzyme may be important in producing a useful
clinical effect. Enzyme inhibiting processes may be divided into two main
classes, reversible and irreversible, depending upon the manner in which the
inhibitor is attached to the enzyme. Reversible inhibition occurs when the
inhibitor is bound to the enzyme through a suitable combination of Van der
Vaals’, electrostatic, hydrogen bonding, and hydrophobic attractive forces.
Reversible inhibitors may be competitive, noncompetitive, uncompetitive, or of
mixed type. During irreversible inhibition, after initial binding of the
inhibitor to the enzyme, covalent bonds are formed between a functional group on
the enzyme and the inhibitor. This is the case, for example, for the
active-site-directed inhibitors (affinity labelling).
The inhibitors used in therapy must possess a high specificity towards the
target enzyme, since inhibition of closely related enzymes with different
biological functions could lead to a range of side-effects. There is a very
large area of enzyme targets as illustrated in Table. For example, dihydrofolate
reductase (DHFR) catalyses the NADPH-linked reduction of dihydrofolate to
tetrahydrofolate. The tetrahydrofolate are cofactors for the biosynthesis of
nucleic acids and aminoacids. The reduction of their level induces a limitation
of cell growth. Thymidylate synthase methylated the deoxyuridylate into
thymidylate using 5,10-methylenetetrahydrofolate as a cofactor. This reaction is
the rate-limiting step in the
de novo
synthetic pathway to thymidine nucleotide. Antitumoral effects are
obtained with antifolate compounds.
A
functional HIV protease is required for the production of infective virions;
this key role of the protease in the viral life cycle makes the inhibition of
this enzyme a potential way for therapeutic intervention in the treatment of
AIDS. New strategies for the development of bifunctional inhibitors, which
combine the protease inhibitor and another enzyme inhibitor in one molecule are
under investigation.
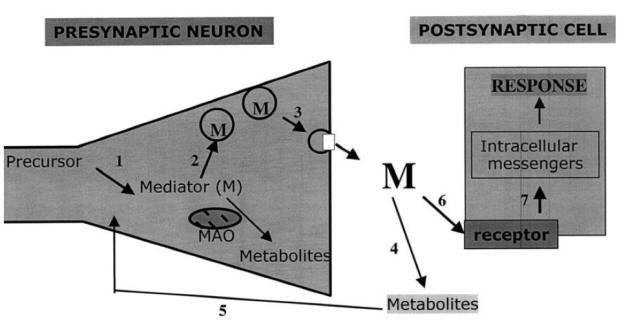
Drugs influencing synaptic transmission by: (1) inhibiting enzymes
that synthesize neurotransmitters; (2) preventing neurotransmitter storage in
synaptic vesicles; (3) blocking the release of neurotransmitter into the
synapse; (4) blocking enzymes that degrade neurotransmitters; (5) blocking
neurotransmitter (or metabolite) re-uptake; (6) binding to the receptor and
either mimicking or blocking neurotransmitters; and (7) interfering with second
messenger activity.
Angiotensin-converting enzyme (ACE) inhibitors are used for the treatment of
high blood pressure, and were designated using the carboxypeptidase structure as
a model for Zn2+
protease action. Captopril is a small, potent, orally available,
dipeptidyl inhibitor of ACE. Acetylcholinesterase (AchE) hydrolyses the
neurotransmitter acetylcholine and yields acetic acid and choline. AchE is a
serine hydrolase inhibited by organophosphorus poisons, as well as by carbamates
and sulfonyl halides which form a covalent bond to a serine residue in the
active site. AchE inhibitors are used in the treatment of various disorders.
In conclusion, the modification by drugs of a precise function can be
achieved in several different ways, acting on the receptors of mediators, on
enzymes or transmembrane exchange processes. This view is illustrated in Fig.
showing a synapse. Future development of drug targets will be based on the
diversity of biological targets, according to the definition of the human
genome, increasing the selectivity of drugs. Recent development in gene drug
delivery systems73 and in antisense oligonucleotide technology might also be
crucial factors in drug development.
Selective illustration of enzymes inhibitors

Protein Kinase Inhibitors
Protein
kinases belong to the family of group transfer phosphorylating enzymes that
transfer a phosphate group from ATP onto amino acid residues of proteins. It is
estimated that there are over 500 of these kinases encoded by the human genome
(the kinome), and this kinase reaction when coupled with a phosphatase
(dephosphorylation) reaction, i.e., reversible phosphorylation of proteins,
offers cells a precise regulatory mechanism to control its differentiation,
maturation, proliferation, apoptosis, and other cellular functions.
The substrate amino acid residues on proteins that are phosphorylated belong
largely to the hydroxyl bearing amino acids such as serine, threonine, and
tyrosine. Hence, these kinases are referred to as either serine/threonine
kinases or tyrosine kinases. Similarly, the phosphatases are referred to as
serine/threonine or tyrosine phosphatases of which the tyrosine phosphatases
predominate. Since these proteins are involved in regulatory functions of the
cell, it becomes intuitive that mutations or aberrations of expression of these
proteins can lead to dysregulation of cellular functions giving rise to tumors
and cancers and other diseases. Indeed, research has shown that of the 518
kinase genes present in the human genome, 244 map to disease loci and cancer.
Moreover, targeting these enzymes with inhibitors would be a way to selectively
target cancers without the noxious side effects seen with conventional
anticancer drugs such as the alkylating agents or antitumor antibiotics.
Research over the last decade has provided this as a rationale to develop
selective (targeted) anticancer therapy where such kinases which manifest
themselves in certain cancers have been specifically targeted for inhibition
resulting in dramatic declines of cancer cells and greater survival times for
the patient.
Examples of such targeted anticancer drugs have involved primarily the
development of the tyrosine kinase inhibitors (TKIs) several of which are FDA
approved; however, more recently, inhibitors of serine/threonine kinases have
entered the clinical market while inhibitors of the tyrosine phosphatases are in
development.
Tyrosine Kinase Inhibitors
The tyrosine
kinases are a group of enzymes responsible for signal transduction and
intracellular signaling functions many of which are involved in cell
differentiation and proliferation. They can be divided into two major types
depending upon where they act in the cell, viz. receptor tyrosine kinases (RTK),
a membrane spanning protein having an extracellular ligand binding domain and an
intracellular catalytic (kinase) domain involved in the transduction of
extracellular signals from the membrane to the cytoplasm, and the nonreceptor
tyrosine kinases involved in cytosolic signaling events.
Inhibitors for these have been developed for both types and have shown excellent
and selective activity in cancers manifested by aberrant expression of these
proteins. The design for selective inhibitors has been based on determining
important binding regions to the protein. The kinase domain has a C-terminal
domain linked via a “hinge region” to the N-terminal domain, and structural
studies have indicated that ATP is known to bind to the backbone of this hinge
region (ATP binding pocket). These kinases all have an “activation loop” which
contains a tyrosine residue (Tyr393), the major phosphorylated residue that
allows switching the kinase from inactive to active forms and allowing for ATP
binding. The large majority of the TKIs bind to this region via H-bonding. Areas
of the protein that have been identified as important for the function of
kinases include a glycine-rich loop (G-loop), ATP binding pocket, a gate-keeper
residue (an amino acid preceding the hinge region), and the DFG activation
motif. Most of the inhibitors that have been developed tend to bind to the ATP
binding pocket and have been classified based on their binding motif. The Type I
inhibitors bind the “DFGin” motif of the activation loop (the active form of the
kinase, where the kinase is poised for the phosphoryl transfer) and is a more
conserved region, while the Type II inhibitors bind the inactive (DFG-out) motif
which is a region of less conserved residues but would allow for greater
specificity. Type III and Type IV inhibitors bind to regions outside of the ATP
binding pocket (distal sites) and are classified as allosteric inhibitors. The
kinase-ligand interaction fingerprints and structures (KLIFS) is a useful
database that identifies the binding pocket for Type I to Type IV inhibitors
which includes the gatekeeper residues for various kinases and is often used to
aid medicinal chemists in designing new kinase inhibitors.
The
development of a tyrosine kinase inhibitor (TKI) to selectively treat a cancer,
chronic myelogenous leukemia, CML, was the impetus leading to the large number
of presently available TKIs. CML in the majority of patients is due to a
reciprocal translocation of chromosomes 9 and 22 resulting in a fusion of the
abl (Abelson leukemia virus) gene of chromosome 9 to the bcr
(breakpoint cluster) gene of chromosome 22 leading to the bcr-abl fusion
gene (the Philadelphia chromosome). While the abl gene normally produces
a nonreceptor tyrosine kinase whose activity is highly regulated, the bcr-abl
fusion gene produces a tyrosine kinase that is constitutively active and
whose activity is required for the transformation of cells to become malignant.
The knowledge of this direct correlation between expression of the abnormal
fusion protein and CML allowed for the development of specific inhibitors for
this protein and other such dysregulated kinases that are overexpressed in many
cancers. Using a high-throughput screening program to develop inhibitors for
receptor tyrosine kinases as a possible treatment for such cancers,
2-phenylaminopyrimidine (PAP) became a lead compound. Further structure activity
optimization and refinement of this lead led to Imatinib, the first targeted
drug for treatment of CML.
Note, the structure optimization to imatinib included addition of a pyridine, a
methyl as well as a benzamide to enhance the potency of the basic PAP nucleus.
The piperazinyl functionality helped to increase water solubility allowing for
better
“druglike” properties. Imatinib also proved to be inhibitory in many other
cancers with overexpressed kinases, such as gastrointestinal stromal tumors
(GIST) (which overexpress c-kit), myelodysplastic diseases associated
with plateletderived growth factor receptor (PDGFR) as well as in Philadelphia
chromosome-positive adult lymphoblastic leukemia.
X-ray
crystallographic studies with imatinib co-cyrstallized with the TK expressed
from abl showed that imatinib binds to the ATP binding site of the
protein in its inactive conformation (DFG-out form—Type II), and this binding
prevented the kinase from achieving its productive binding conformation with
ATP.
Studies showed that with the protein-bound imatinib, Tyr393 was not
phosphorylated and the conformation of this activation loop in the
nonphosphorylated protein changed to that resembling substrate (ATP) being bound
to the kinase. In this way, the altered geometry brought about by imatinib
binding to the protein prevented the protein from binding its true substrate,
ATP.

Resistance develops to
imatinib due to mutations (in the hydrophobic pocket, gate keeper residue being
mutated to a larger residue) that prevent access of imatinib to the protein in
the off state, thus allowing for the kinase to bind ATP and cancer to progress.
TKIs dasatinib and bosutinib have also been developed
to bind the kinase in its “on” (active conformation, Type-I) where the drugs can
access hydrophobic regions in the ATP binding pocket as well as drugs that can
bind both “on” and “off” forms—dual mode inhibitors which are more potent than
imatinib.
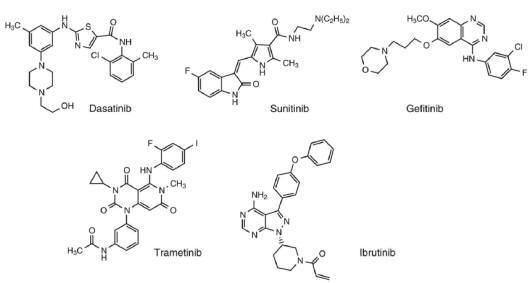
It has been
more than 15 years since the introduction of imatinib, and today many other TKI
drugs have been developed for related tyrosine kinases such as epidermal growth
factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), and
vascular endothelial growth factor receptor (VEGFR). The inhibitors make use of
differences in the variable region of the protein surrounding the ATP binding
pocket which allows for specific binding interactions with the various
functionalities present on the individual inhibitors. Resistance to these
inhibitors manifest themselves due to mutations to these variable regions on the
protein as well as to cellular efflux pumps being activated. While the vast
majority of these clinically used drugs are reversible inhibitors of the Type I
and II, more recently Type III inhibitors have been introduced in the clinic.
Furthermore,
irreversible inhibitors of Type I which employ the “Michael acceptor”
functionality to irreversibly alkylate an active site cysteine residue have also
been introduced. Examples of these inhibitors such as dasatinib (Type I for
imatinib resistance), sunitinib (Type I, inhibitor of VEGFR), gefitinib (Type I,
inhibitor of EGFR), trematinib (Type III, inhibitor of MEK), and the
irreversible Type I inhibitor, Ibrutinib with the α, β unsaturated system
(Michael acceptor) are shown in
Figure.
Serine/Threonine Kinase Inhibitors
The
serine/threonine kinases are a family of enzymes that phosphorylate the hydroxyl
groups of serine and threonine present on proteins. There are a number of such
serine/threonine kinases that are important regulators of cell proliferation and
survival and whose dysregulation often lead to cancer and tumorigenesis. Protein
kinase C is perhaps the most well-studied system, whose activation results in
the formation of diacylglycerol (DAG), phosphatidylinositol-3,4,5-triphosphate
(PIP3), and concomitant increase in intracellular calcium, leading to various
signal transduction events in the cell through activation of the
mitogen-activated protein kinase (MAPK) family. Furthermore, other
serine/threonine kinase proteins such as PI3K (phosphoinositide-3-kinase), Akt
(protein kinase B), and mTOR (mammalian target of Rapamycin) are part of an
intracellular signaling pathway that is important for regulation of cell
proliferation, migration, and survival. Initial events leading to cell
proliferation via the PI3K-Akt-mTOR have been shown to begin with growth factor
or hormonal activation of a receptor tyrosine kinase which leads to activation
of PI3K. This activation results in the release of PIP3 which in turn leads to
phosphorylation and further activation of Akt and mTOR. Inhibitors of mTOR
include rapamycin (sirolimus) and its derivatives, everolimus and temsirolimus.
Rapamycin is a macrolide originally isolated from a microbe on Easter Island and
primarily used as an immunosuppressant drug while its derivatives, everolimus
and temsirolimus, have been used to treat renal cell carcinoma.

Additional
development of molecules to specifically inhibit the PI3K-Akt pathway include
copanlisib, which was recently introduced to treat follicular lymphoma, and
duvelisib, a first-in-class inhibitor of PI3K that has recently received FDA new
drug application (NDA) status to treat chronic lymphocytic leukemia (Fig.).
Additional serine/threonine kinase function is also observed during mitosis,
which is a highly regulated process with multiple checkpoints encountered during
the chromosomal segregation stage of cell division. The phosphorylation of
specific serine/threonine residues by these mitotic kinases (also known as
Aurora A and Aurora B kinases) serves as important checkpoints during mitosis.
These Aurora kinases interact with many proteins, including tumor suppressors
and activators from the mitotic entry stage all the way to cytokinesis. It is
not surprising then that they are overexpressed in many tumors (breast, colon,
gastric, ovarian, and pancreatic) and thus have become an attractive target for
anticancer drug development.
Examples of some of these inhibitors include
alisertib, danusertib, and a phosphate-based prodrug currently under
development, barasertib (Fig.).
Other
serine/threonine kinases include the RAF proteins which are involved in the MAPK
activation cascade during cell growth. B-RAF is one member of the RAF family
where mutations (specifically the V600E) have resulted in dysregulation of the
cascade leading to many cancers. Specific clinically used inhibitors of the
V600E B-RAF mutated protein include vemurafenib and dabrafenib (Fig.)
used to treat metastatic melanoma.

ATPase Inhibitors
The transmembrane proteins establishing these gradients are
transporters such as the Na+–K+ ATPase pumping three Na+ out and two K+ into the
cell while consuming one ATP molecule. Other transporters are the Ca2+-ATPases
pumping Ca2+ out of the cell or into the endoplasmatic reticulum, and secondary
active transporters such as the Na+–Ca2+ exchanger not using energy themselves,
but exploiting the ion gradients created by the ATPases. The transporters
typically move 0.1–10 ions/ms each, they show saturation kinetics like enzymes,
and they slowly build up the ion gradients.
The sesquiterpene lactone, thapsigargin which is structurally
unrelated to ryanodine, also interacts with an intracellular calcium mechanism.
Thapsigargin has become the key pharmacological tool for the characterization of
the sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA). Thapsigargin effectively
inhibits this ATPase, causing a rise in the cytosolic calcium level which
eventually leads to cell death. Although the SERCA pump is essential for all
cell types, attempts to target thapsigargin toward prostate cancer cells have
been made based on a prodrug approach.
Gastroesophageal reflux disease (GERD) is one of the most common GI disorders
and affects a significant number of people in the United States and worldwide.
GERD is typically associated with the esophageal mucosa being continuously
exposed to gastric secretions. This often occurs when the lower esophageal
sphincter relaxes and the duration of esophageal acid exposure is considerably
prolonged. Typically, when gastric secretions enter the esophagus, innate
clearing mechanisms limit the duration of exposure by rapidly pushing the
refluxed content into the stomach. Additionally, bicarbonate-rich secretions by
esophageal glands help neutralize the residual acid trapped within the mucosa.
Since GERD is associated with gastric acid reflux and associated symptoms, most
treatment options are directed toward reducing the acidic nature of the
refluxate. An effective therapeutic option should heal esophageal damage while
providing symptomatic relief. Current therapy for acid reflux–related GI
diseases focuses on inhibiting biological pathways that directly or indirectly
play a role in stomach acid secretion. Two such pathways involve inhibition of
the histamine H2 receptor and inhibition of the proton pump (H+/K+-ATPase) in
the stomach. Selective muscarinic receptor antagonists and prokinetic agents can
also be used for the treatment of GERD.
Proton Pump Inhibitors
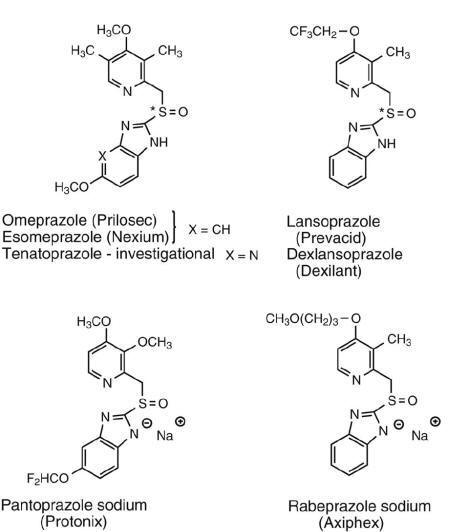
A second class
of drugs that are commonly and more frequently used in the treatment of peptic
ulcer is PPIs.
This class of drugs covalently inhibits the gastric H+/K+-ATPase
that is responsible for secretion of stomach acid in parietal cells of the
gastric mucosa.
The gastric proton pump is very similar to Na+/K+-ATPase,
and also similar in structure and function to the H+/K+-ATPase found in
osteoclasts, which play an important role in bone resorption. PPIs enable
healing of peptic ulcer, erosive esophagitis, GERD, GERD-related laryngitis,
Barrett’s esophagus, and Zollinger-Ellison syndrome, as well as the infection
caused by H.pylori, the latter in combination with antibiotics.
Since the H+/K+-ATPase–mediated process is the final step in gastric acid
secretion, inhibiting this enzyme is considered the most effective approach to
acid suppression. PPI discovery began with the early investigation of
timoprazole. This compound is a pyridyl methyl sulfinyl benzimidazole, which is
the conserved pharmacophore for subsequently developed PPIs. Based on its acid
dependent activity, it was identified as an acid-activated prodrug. Omeprazole
was later synthesized and, in 1989, became the first drug of this class
available for clinical use.
Other clinically available PPIs include lansoprazole,
dexlansoprazole, esomeprazole, pantoprazole sodium, and rabeprazole sodium (Fig.).
Dexlansoprazole and esomeprazole are enantiomerically pure forms of lansoprazole
and omeprazole, respectively. Omeprazole and lansoprazole are both also marketed
as racemic mixtures.
Structural Features and Mechanism of Action of PPIs
The
2-pyridylmethylsulfinylbenzimidazole motif is conserved in all members of the
PPI family, as it is necessary for bioactivity through acidcatalyzed
decomposition to the reactive sulfenic acid and sulfenamide structures.
Typically, the structural modifications are done on the pyridine ring or at the
benzo component of the benzimidazole to generate drugs with differing levels of
acid stability, which impacts duration of proton pump inhibiting action. The
investigational drug, tenatoprazole is slightly different in that it has an
imidazopyridine isostere, instead of the traditional benzimidazole scaffold.
PPIs are weak bases with a pyridine pKa between 3.8 and 4.9, which
enables them to selectively accumulate through ion trapping in the stimulated
parietal cell (pH∼1.0). This acidic
environment-selective accumulation of PPIs is an important property that
contributes to their selectivity and activity. As mentioned, PPIs are prodrugs,
and acidcatalyzed activation generates the active form of the drug. The active
form has electrophilic sulfenic acid and sulfenamide sulfur atoms capable of
reacting with the thiol group of a Cys residue of the ATPase, forming a covalent
adduct.
Covalent inhibition of the proton pump results in inactivation of the catalytic
function of this enzyme.
The chemical mechanism of activation of omeprazole and subsequent
covalent interaction is described in
Figure.
All PPIs follow a similar mechanism of
action. The imidazole ring N3, an exceptionally weak base with a pKa
<0.8, is first protonated under the very low pH of the parietal cell through
proton transfer from pyridine N1. Nucleophilic attack by the unionized pyridine
nitrogen at benzimidazole C2 (made electron deficient by cationic N3) generates
a highly electrophilic and unstable electrophilic spiro intermediate, which
rearranges to generate the sulfenic acid intermediate that preferentially forms
the inactivating disulfide bond with the proton pump Cys (Fig).
The following step involves loss of water and the formation of a reactive
sulfenamide intermediate, which can also covalently modify the Cys thiol of the
proton pump.
Within the
proton pump, several different Cys residues are potential sites for covalent
modification. The different substituents on the pyridine or benzimidazole ring
of PPIs determine which Cys residue the drug preferentially reacts with, which
subsequently determines the permanency of the covalent attachment, as discussed
below. Electron donating groups on the C5-position of benzimidazole enhance
reactivity by increasing the extent of benzimidazole N3 protonation and the
strength of the δ+ charge on the carbon at the C2 position of benzimidazole
moiety. Electron-donating groups at the C4 position of the pyridine ring
increase the nucleophilicity of pyridine nitrogen, enhancing the rate at which
N1 attacks electrophilic benzimidazole C2. Conversely, electron-withdrawing
groups at either site have been shown to decrease reactivity.

All PPIs can
react with the readily accessible Cys813 of the ATPase. It hasbeen observed that
activated molecules of omeprazole can bind to either Cys813 or Cys892 of the
proton pump.
Similarly, lansoprazole is known to covalently modify Cys813 or Cys321.
Pantoprazole, the most sluggish-reacting of the PPIs, can react with either
Cys813 or the more deeply recessed and less accessible Cys822. Although PPIs are
covalently bound to the proton pump, the interaction with accessible Cys
residues can be reversed by reduced glutathione (GSH). The disulfide bond formed
between the drug and receptor can be cleaved by this endogenous reducing agent,
regenerating the essential Cys thiol of the pump and prompting the excretion of
the drug fragment as a glutathione conjugate. Due to low concentration of GSH in
the body, regeneration of proton pumps inhibited through Cys813 is incomplete.
Pantoprazole binding is found to be less susceptible to GSH-mediated removal.
These observations suggest that modification of Cys813 is more easily reversed,
providing a fast phase of recovery, while modification of Cys822 is difficult to
reverse, as GSH may not easily reach this site.
Since the
activation and rearrangement of PPIs take place at a strongly acidic pH, all of
the oral formulations of PPIs have been developed to be acid-stable. This allows
for better dissolution and absorption of PPIs (typically enteric-coated
formulations) from the intestines. Both lansoprazole (enteric-coated) and
omeprazole (either enteric coated or formulated with NaHCO3) are available in
granular form. This allows for the drug to be absorbed readily with minimal drug
destruction in the stomach. Rearrangement selectively occurs in the acidic
environment of the canaliculus within parietal cells after the drug is absorbed
from the intestine and delivered to that target site. Typically, the half-life
of PPIs is relatively short (∼1
hr); however, the duration of action is long (∼20-48 hr) because of their ability to irreversibly inhibit the
proton pump.
Benzimidazole
N1 is typically nonionizable at physiological pH and considered neutral in
nature. However, as noted in
Figure, benzimidazole N1 within PPIs is slightly
acidic due to the electron withdrawing property of the attached sulfinyl moiety.
Due to this change in acidity, PPIs can be made into salt forms when
benzimidazole N1 ionizes (loses proton) through treatment with strong base and
becomes paired with the resultant metal cation (e.g., Na+ or Mg2+). Due to this
property, some of the PPIs are marketed in the salt form, which providesimproved
aqueous solubility.
Although the proton pump inhibitors, such as omeprazole,
lansoprazole, pantoprazole, rabeprazole, esomeprazole and tenatoprazole, were
not originally developed as prodrugs, they provide a good example of
site-selective prodrugs. Due to the basic pyridine group (pKa
of omeprazole is 3.97), these drugs are protonated and accumulated in the acidic
secretory parietal cells (Figure). In the acidic conditions of parietal cells
(pH of 1–2), prazoles undergo spontaneous chemical reaction to their active
sulfenamide metabolites followed by their irreversible binding to a cysteine
group of H+/K+ ATPase. This irreversible binding inhibits the ability of
parietal cells to secrete gastric acid. The fact that proton pump inhibitors are
only effective on H+/K+ ATPases, which contain highly acidic compartments that
nongastric H+/K+ ATPases lack, corresponds their excellent safety profiles.
Therefore, proton pump inhibitors are converted to their active species only
under highly acidic conditions at their site of action.
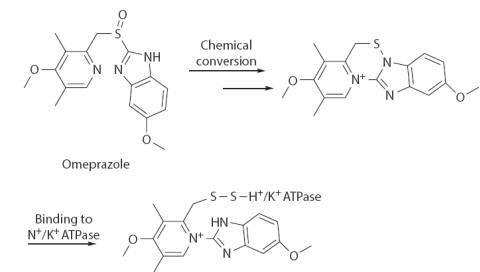
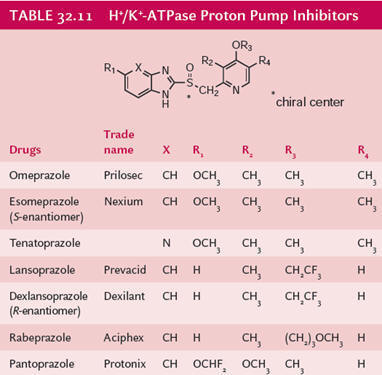
Binding Site and Molecular Interactions
The binding site provides a base for the interaction of two
molecules and describes the ability of the receptor to form bonds with other
substances. Based on the bound molecule, the binding site can be protein–protein, protein–nucleic acid,
protein–carbohydrate,
protein–lipid, and protein–small molecule
binding sites. The binding of the drug molecule to plasma proteins (albumin,
lipoproteins, and globulins) is a major determinant of drug distribution.
Protein–Drug
Interactions
DNA and protein are molecules that show interaction with other
small molecules such as substrate, drug, or other ligands. Proteins are an
important molecule in all living cells and play an essential role in various
cellular processes in the form of enzyme, hormone, and receptor. Each protein
performs a specific function that is
governed by its 3D structure. Protein–ligand interactions
are vital for all biological processes that occur in living organisms. The
function of a protein depends upon the specific sites that are
designed to bind with a specific ligand molecule.
Ligand-binding interactions can alter the protein conformations and its
function. To perform its function properly, binding of a protein with other
molecules should be very specific. A drug is a small
organic molecule, which binds to the receptor and forms a protein–drug complex and
controls the function of biological receptors. Binding can be of two types:
intracellular or extracellular. Based on the drug binding mechanism, drug
binding may be of reversible or an irreversible type.
Reversible Binding
In reversible binding, usually, the drug binds the proteins with
weaker chemical bonds such as hydrogen bonds, hydrophobic bonds, ionic bonds,
and van der Waals interactions. The binding of drugs to plasma protein is a
reversible process.
Irreversible Binding
In the case of irreversible binding, a drug or inhibitor
permanently binds with the binding site of the drug target. Irreversible binding
of drugs rarely takes place. As a result of covalent bonding or strong force of
interaction between drug and target protein, the event of carcinogenicity or
cellular toxicity takes place.
Factors Affecting Protein–Drug
Binding
(a) Drug-related factors: It includes physicochemical
characteristics of the drug, concentration of drug in the body, and affinity of a drug.
(b) Protein/tissue-related factors: It includes physicochemical
characteristics of protein or drug and concentration of protein or drug.
(c) Drug interactions: It contains allosteric changes in a protein
molecule, competition between drugs to occupy the binding site, and competition
between drug and biological components.
(d) Patient-related factors: It includes the age of the patient,
inter-subject variabilities such as due to genetics, environmental factors, and
disease states.
Altogether, more protein binding disturbs the absorption and also
decreases the distribution and metabolism of drugs.
Role of Water Molecules
In the last 10–15 years, the significance of water molecules in drug design and protein structures has
become of extensive interest. Traditionally, water molecules play two crucial
roles in ligand binding. Water molecules stabilize a protein–ligand complex by
contributing hydrogen bond interaction between a ligand and a protein. The
second role is that water can be displaced by ligands on binding with the target
protein. The role of the water molecule in binding interaction of a ligand with
the active site of the target protein can be studied using the molecular
dynamics simulation. Slight changes in water-based hydrogen bonding networks
affect ligand–protein interaction
energies and show the effect of solvation or water molecule on the binding.
Water molecules also determine the binding or rejection of ligand to the binding
site of proteins. Water molecules can mediate to direct interactions or may
cause an effect of electrostatic screening.
Drug–Nucleic
Acid Interactions
Nucleic acids are the carrier of genetic information, and hence
they are an important molecule for disease prevention. Nucleic acids are
targeted for various diseases, including various types of cancer. DNA, the
carrier of genetic information in humans, is mutated in various diseases, which
often result in gene expression alteration. The structures of DNA can be used
for designing small molecules to regain the gene expression pattern. The small
molecules which bind to DNA can be categorized into two major classes: (1)
covalent binder and (2) non-covalent binder. The non-covalent binders can
further be classified into major groove
binders, minor groove binders, and intercalators. On the other hand, RNA itself
regulates various activities from catalysis to gene expression, which makes RNA
a suitable target for binding. Since the structure of RNA is highly variable,
designing a small molecule against them is a challenging task. The advancement
in technology and growth in the structures of protein–nucleic
acid complexes help the
computational tools to predict the binding of small molecules with nucleic
acids.
Receptor Pharmacology
1.
Agonist: The drug which show affinity for the receptor and activates the
receptor to produce regular physiological function are called as agonist.
Agonist molecules mimics the effects of endogenous mediators.
(a)
Full agonist: The drug with similar pharmacological potential (similar
affinity strength) compared with the natural ligand is called as full agonist.
(b)
Partial agonist: The drug with less pharmacological potential compared
to the natural ligand (irrespective of concentration) is called as partial
agonist.
2.
Inverse agonist: The drug showing affinity for the receptor and producing
opposite pharmacological activity is called as inverse agonist. The molecule
beta-carboline and diazepam shows similar affinity for the benzodiazepine
receptor. But, beta-carboline produce excitation of the receptor and diazepam
produces inhibitory effect.
3.
Antagonist: The drug which offer affinity for the receptor, but do not
activates the receptor are called as antagonist.
(a)
Competitive antagonist: The antagonist drug, which binds to the binding site
of the natural ligand with similar affinity (blockade of natural ligand) and
produce no biological response is called as competitive antagonist.
(b)
Non-competitive : The antagonist drug which binds to the receptor on the
site other than the binding site specific for agonist (allosteric binding site)
is called as non-competitive antagonist. It induces the newer conformational
change (different from the agonist conformation), hence natural ligand and
agonist molecules do not find receptor (no biological response).
Kinetic analysis of ligand–receptor interactions
Theoretical background
Considerable evidence suggests that 7TM receptors can exist in two different
coexisting conformational states: an ‘inactive’ state R and an ‘active’ state
R*. The relative proportion of R and Rp
will depend on both the actual receptor–effector system and the
presence of ligands (L) having affinity for the receptor, i.e. able to form
complexes with R (LR) and R*
(LR*). Only R*
and AR*
can couple with G-proteins to induce a cellular response (e.g.
increase in cAMP levels). If, to simplify, the reversible interaction between
the receptor and the G-protein is omitted, the minimal scheme depicting the
relationship between A, R, R*, LR and
LR*
can be written as follows:
Scheme 1

where:
(1)
g
is an intrinsic allosteric constant (g
=
R*/R), reflecting the relative proportions of R and R*
forms which coexist in the absence of any ligand L.
(2) Ka and
a
Ka are
the association constants of L for R and R*,
respectively. These constants actually measure the affinity of the ligand L for
the R and R*
forms of the receptor. The factor
a
denotes the differential affinity of L for R*. This means that L, along with
g, can modify the relative proportions of R, R*, LR and LR*
(see Scheme
1).
Two main experimental situations can be observed (Fig.):
(1) In the absence of any ligand L, a cellular response can be easily measured.
In such constitutively active systems (e.g. native or mutated receptors, cells
highly transfected with a particular cloned receptor) a sufficient proportion of
R*, spontaneously present, binds to the G-protein to induce a cellular
response (high basal level). In this case,
ligands L are classified according to the value of the
a
constant, as full agonists and partial agonists
(a
>
0), neutral antagonists (a
=
0) and
inverse agonists (a
<
0) (Fig.). Thus, the higher the absolute value of the
a
constant, the higher the cellular response. The
a
constant actually measures the intrinsic
efficacy—positive, null or negative — of a particular ligand L for a given
receptor–effector system. In this respect, a neutral antagonist with zero
efficacy, does nothing to the receptor but bind to it and therefore preclude
activation of the receptor by an agonist. As pointed out by Kenakin8 efficacy is
‘the property of a drug that modifies subsequent interaction of the 7TM receptor
with other membrane proteins’. The inverse agonism patterns depicted in Fig.
have been observed with
b-adrenergic ligands on basal adenylyl cyclase
activity mediated by a constitutively active mutant of the
b2-adrenergic receptor9 or on Sf9 cells overexpressed with
b 2 wild-type receptors as well.
(2) A low basal level of activity is measured in the absence of any ligand L. In
such quiescent systems (e.g. in general native wild-type receptors present on
cellular or tissue membranes) inverse agonists cannot be revealed since they
behave as neutral antagonists. In contrast, as in the former situation, full and
partial agonists will induce a cellular response the magnitude of which depends
on their efficacy (Fig).
Evolution of the cellular response as a function of the ligand and
the receptor–effector complex. (A) Constitutively active systems. (B) Quiescent
systems.
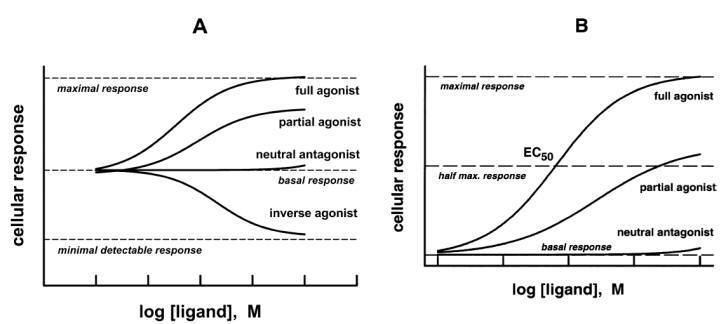
Quantitative measurement
As far as affinity and efficacy of drugs are concerned in their therapeutic
activity in humans, the correct measurement of these parameters using
well-chosen and reliable methods is highly important for drug discovery. These
methods mainly, but not exclusively, concern radioligand-binding techniques and
specific functional assays.
Radioligand binding
The simplest assumption about the reversible formation of a ligand–receptor
complex (see left part of Scheme
1) is
that it may be expressed according to the mass-action law, as the following
chemical reaction:
![]()
where L is the ligand,
k1 and
k-1 are
the rate constants for association and dissociation of the LR complex. Square
brackets signify the concentration of the entity they enclose. At equilibrium
(or steady state), the rates of association and dissociation of the complex LR
are equal, and:
![]()
The equilibrium binding constant may then be defined as dissociation constant (kd) such as:
![]()
The total number of receptors (RT) is the maximum number of receptors on which
the drug is able to bind specifically. This finite number, also termed
Bmax, is the sum of the receptors engaged in
forming the complex (LR), plus the free receptors (R), such as:
![]()
From equations (3) and (4) after rearranging:
![]()
If we now define LR as bound ligand
=
B, and L as free ligand
=
F, from
equation (5):

Equation (7) is the Scatchard equation. Receptor labelling studies involve
binding of a radioactive form of the ligand to receptor preparations of target
tissues. Radiolabelled ligands — ideally neutral antagonists that are devoid of
efficacy for the receptor used — must be pure, stable and have a high
radiochemical specific activity. The most commonly used isotopes are tritium [3H] and radioactive iodine [125I].
Radioactive ligand and receptor preparations are incubated in appropriate
conditions: receptor concentration, temperature, pH and ionic strength. At the
end of the incubation period, while the binding reaction is at equilibrium,
bound and free ligands are promptly separated using filtration or
centrifugation.
Experimental determination of
kd and
Bmax for a given radioligand and receptor
preparation requires incubation of various concentrations of the radioligand
with a fixed concentration of receptors: this is the so-called saturation
experiment. Such experiments can be performed in the absence (control) or
presence of various concentrations of non-radioactive (‘cold’) ligand or drug —
also termed displacer — able to compete with the radioligand for the same
receptor. Finally, interaction of displacers with a given receptor can also be
studied by incubating various concentrations of the displacer with fixed
concentrations of radioligand and receptors: this is the so-called displacement
experiment (Fig.). In all cases, the non-specific binding is measured by the
bound radioactivity remaining in the presence of a large excess of cold ligand.
In most cases, data generated from a saturation experiment are analysed
according to equation (7). When the radioactive ligand interacts with a single
population of non-interacting receptors, the Scatchard-plot, i.e. B/F vs B,
leads to a linear curve.
Kd is estimated as the negative reciprocal of the
slope of the line of best fit, and
Bmax by the abscissa intercept of the line (Fig.). The reciprocal of
Kd measures the
association constant Ka (see Scheme 1) of the radioligand for the
receptor. Thus, for a given ligand–receptor pair, the smaller the
Kd (generally 0.1–10 nmol l-1
or nM) the higher is its
affinity.
Bmax is
expressed as pmol or fmol per mg tissue or protein. A downward curvilinear
Scatchard plot may indicate that there is more than one type of binding site,
and that the different types differ in their dissociation constants for binding
with the radiolabelled ligand under investigation. Alternatively, the binding of
ligand to its receptors may inhibit further binding and hasten dissociation from
the receptor (e.g. insulin receptor). This latter phenomenon is called
negative cooperativity.
Positive cooperativity
also
occurs, i.e. the binding of ligand facilitates the binding of subsequent
molecules to the receptors (e.g. nicotinic receptor). In this case, the
Scatchard plot is upward concave. Cooperativity generally implies that the
ligand behaves as an allosteric modifier that induces a conformational change in
the structure of the receptor protein.
Analysis of ligand–receptor interactions. (A) Scatchard plot in
absence (control) or presence of two different displacers. X and Y are
competitive and non-competitive displacers, respectively. (B) Displacement
curve.
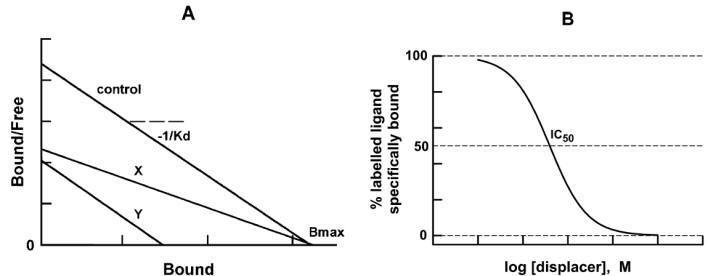
When the saturation experiment is performed in the presence of a displacer, the
line of best fit of the Scatchard plot can be modified in a manner that depends
on the type of receptor interaction exhibited by the displacer. Two main cases
exist: (1) decreased slope and unchanged
Bmax, the displacement is of the competitive type; (2) unchanged slope
and decreased
Bmax, the
displacement is of the noncompetitive type (Fig.A). Intermediate cases where
both the slope and
Bmax are
modified also exist. Data generated from a displacement experiment are generally
fitted by a sigmoid curve termed
displacement
or
inhibition curve,
i.e. percentage radioalabelled ligand specifically bound vs log [Displacer] (in
mol 1-1 or M) (Fig. B). The abscissa of the inflexion point of the curve
gives the IC50 value, i.e. the concentration of displacer that displaces or
inhibits 50% of the radioactive ligand specifically bound. IC50 is a measure of
the
inhibitory
or
affinity
constant
(Ki) of
the displacer for the receptor. IC50 and
Ki are linked as follows: if the displacement is of the competitive
type, then,

Forces involved in
drug-receptor complexes
According to the receptor theory of drug action, the common event in the
initiation of pharmacological responses is the formation of a complex between
the drug molecule and its site of action. Since most pharmacological responses
are mediated through receptors, recognition of the more mobile drug molecules by
the cellular receptor is the critical element determining the specificity of the
response. There must be some forces that not only attract the drug to its
receptor, but also hold it in combination with the receptor long enough to
initiate the chain of events leading to the effect. Thus the combination of
various chemical bonds is of great interest to drug potency.
·
Hydrophobic binding plays an important role in stabilizing the
conformation of proteins and in the association of hydrophobic structure between
the drug and the receptor.
·
Hydrogen bonding, which is strongly directional, has considerable
importance in stabilizing structures by intramolecular bond formation. The
formation of such bonds between a drug and a receptor can result in a relatively
stable and reversible interaction. Such bonds are also involved in the
maintaining of the tertiary structure of the receptor macromolecule and are
thought to be involved in the specificity and selectivity of drug–receptor
interaction.
·
Drug–receptor interaction often involves charge transfer complexes
formed between electron-rich donor molecules and electron-deficient acceptors.
The angiotensin-converting enzyme (ACE) inhibitor, captopril
interacts with ACE through electrostatic interactions.
(a) The carboxylic acid group (negatively charged, acidic) of the
captopril binds to positively charged glutamate and lysine.
(b) The amide group of captopril establishes dipole-dipole
interaction (hydrogen-bond) with histidine.
(c) The mercapto group makes dipole interaction with glycine.
(d) The ACE tyrosine residue bonds with carboxylic group of
captopril through hydrogen-bond.
Electrostatic interaction between the captopril and angiotensin-converting
enzyme (ACE).

·
Ionic bonds which are very ubiquitous are of importance in the
actions of ionizable drugs since they act across long distances. The formation
of an ionic bond results from the electrostatic attraction that occurs between
oppositely charged ions. Most receptors have a number of ionizable groups (COO2, OH2, NH3þ) at physiological pH that are available for the binding with charged
drugs.
Non-covalent interaction: Interaction between the adrenaline and adrenergic
receptor through electrostatic, hydrogen-bond, van der Waals and hydrophobic
interactions.

Electrostatic interactions are grouped based on the type of
charge present in the molecules/atoms.
1. Charge-charge interaction
2. Charge-dipole interaction (ion-dipole)
3. Dipole-dipole interaction
4. Hydrogen bond interaction
5. Van der Waals interaction
6. Hydrophobic interaction
Charge-Charge Interactions
The deprotonation of acidic side chain groups present in aspartic
acid and glutamic acid provides anionic environment. The protonation of basic
side chain groups of lysine, arginine and histidine provides cationic
environment.
Bond Strength:
The bond strength of this type of interaction is in the range of
-20 to -40 kJ/mol.
Salt bridge:
The carboxylic group of glutamic acid and the ammonium group of
lysine (charge centers are separated by 4 Å) makes ionic interaction. This
interaction is known as a salt bridge and is essential for the bio-active
conformation of the receptor. The ligand bound receptor establishes salt bridge.
Charge-Dipole (Ion-Dipole) and
Dipole-dipole Interaction
Dipole moment:
It indicates the movement of electrons along the bond. The higher
electronegative atom draws electrons towards it and becomes electron rich centre
(bears partial negative charge;
d˜). The other
atom experiences electron deficiency and bears partial positive charge (d+).
The permanent dipoles of the
carbonyl and amide groups of the peptide are structural determinants. The
dipole-dipole (permanent) interactions are much weaker than charge-charge
interactions. A dipole on one group induces dipole on neighbouring group and
establishes much weaker dipole-induced dipole interaction.
Bond Strength:
The partial charge of dipole is less than that of an ion; hence
charge-dipole and dipole-dipole interactions are weaker than ionic bonds. In
most cases these interactions provides a
DG of –2 to –20 kJ/mol.
Hydrogen Bond Interactions
A strong electrostatic interaction between the hydrogen atom
attached with a hetero atom and a hetero atom (or electro negative atom) is
called as hydrogen-bond. The hetero atoms include oxygen, nitrogen and sulphur.
The electronegative atoms namely fluorine and chlorine also behave as hetero
atoms. The interaction between weakly acidic (partial positive) hydrogen bond
donor (HBD) group and a hydrogen bond acceptor (HBA) group (partial negative)
generates hydrogen bond.
The hetero atom attached with hydrogen (one or two) are known as
hydrogen bond donors (HBDs). The hydrated hetero atom such as oxygen, nitrogen
and sulphur are examples. A protonated tertiary (3°) nitrogen acts as a strong
hydrogen bond donor (HBD). In biological systems these HBD and HBA are highly
electronegative nitrogen (N) and oxygen (O) atoms (occasionally sulphur atoms).
The heteroatom attached with no hydrogen is known as hydrogen bond acceptor
(HBA). The tertiary (3°) nitrogen can act as a strong hydrogen bond acceptor
(HBA). Each HBD forms interaction with two HBA (reverse is also true) and are
responsible for the macromolecular structure. The hydrogen-bonding is a type of
dipole-dipole interaction. The polarisable
p electron system of aromatic
system acts as weak acceptor and relatively weak acidic groups (-CH) acts as
weak donor groups.
Bond strength: The association energies of hydrogen bonds range from -12 to
-20 kJ/mol. The distance between the HBD and HBA groups are normally in between
2.7 to 3.1 Å.
·
Intramolecular hydrogen bond:
The
hydrogen-bond between different parts of a single molecule are known as
intramolecular hydrogen bond. The intra molecular hydrogen bond generally form
six membered ring structures (rigid) and increases the hydrophobicity behaviour
of molecules. These types of bonds are known as bifurcated hydrogen bond. The
multiple inter molecular hydrogen bonds introduce the conformational stability.
·
Intermolecular hydrogen bond:
The
hydrogen-bond between different molecules is known as intermolecular hydrogen
bond. This kind of hydrogen -bond enhances the drug aqueous solubility.
Van der Waals Interactions
The non-covalent interactions between induced dipoles in
electrically neutral molecules are collectively called as van der Waals force.
The transient dipole of non-polar molecules induces dipole in the neighbouring
group. The two non-polar molecules approach each other (close) due to their
temporary polarizability. The attractive forces that hold non-polar molecules
together in liquid phase are known as London dispersion forces. This type of
forces is most common in interiors of protein (buried) and influences the
conformation of the proteins.
Bond strength:
The bond strength in the range of –2 to –4 kJ/mol (weaker) and is
inversely proportional to the 7th power of distance. In high
molecular weight molecule the summation of these forces provides significant
bonding.
Hydrophobic Interactions
It describes the tendency of non-polar molecules to transfer from
aqueous phase into an organic phase. The association of non-polar drug molecule
with the biological receptors results through hydrophobic interactions. A
non-polar molecule cannot be solvated by water. The water molecules surrounding
non-polar molecules associate to form quasicrystalline structure (ice-bergs).
This facilitates the contact of non-polar part of drug molecules to the
receptor.
Bond strength: The association energies of hydrophobic interactions range from
0.1 to 0.2 kJ/mol.
Aryl-aryl interaction (p-p
stacking): The stacking arrangement of an
electron-poor and electron-rich aromatic ring offers charge transfer.
·
The covalent bonds are less important in drug–receptor interaction.
Since bonds of this type are so stable at physiological temperature, the binding
of a drug to a receptor through covalent bond formation would result in the
formation of a long-lasting complex. Although most drug–receptor interactions
are readily reversible, some drugs, such as anticancer nitrogen mustards and
alkylating compounds form reactive cationic intermediates (i.e. aziridinium ion)
that can react with electron donor groups on the receptor. Covalent bonds are
also seen for example in the case of penicillin, which acylates a transpeptidase
enzyme that is essential to bacterial cell-wall synthesis. In this case a
long-lasting inhibition of bacteria replication is needed. Most drugs, however,
induce a brief formation of a reversible drug–receptor complex.

Types
of molecular interactions and their distance strength.
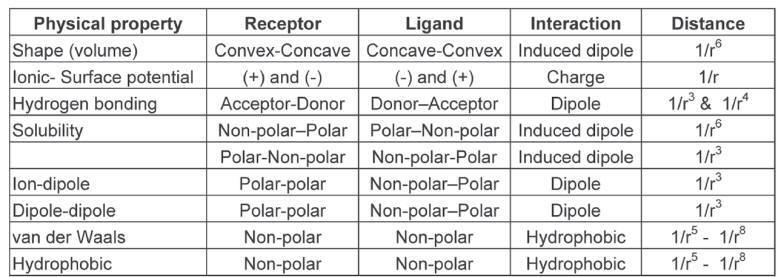
Recombinant versus In Situ Assays
The last decades have had a profound impact on how receptor
pharmacology is performed. As Mentioned in the introduction, receptor cloning
was initiated in the mid-eighties and today the vast majority of receptors have
been cloned. Thus, it is now possible to determine the effect of ligands on
individual receptor subtypes expressed in recombinant systems rather than on a
mixture of receptors in, e.g., an organ. This is very useful given that receptor
selectivity is a major goal in terms of decreasing side effects of drugs and
development of useful pharmacological tool compounds which can be used to
elucidate the physiological function of individual receptor subtypes.
Furthermore, recombinant assays allow one to assay cloned human receptors which
would otherwise not be possible. Most receptors have very similar human and
rodent sequences, but due to the small differences in primary amino acid
sequence there have been cases of drugs developed for rats rather than for
humans, because the compounds were active on the rat receptor but not on the
human receptor.
It should be noted that the use of organ and whole animal
pharmacology is still required. As previously noted, the cellular effect of
receptor activation depends on the intracellular contents of the proteins
involved in, e.g., the signaling cascades. These effects can only be determined
when the receptor is situated in its natural environment rather than in a
recombinant system. In most situations, both recombinant and in situ assays are
thus used to fully evaluate the pharmacological profile of new ligands.
Furthermore, once a compound with the desired selectivity profile has been
identified in the recombinant assays, it is important to confirm that this
compound has the predicted physiological effects in, e.g., primary
nonrecombinant cell lines, isolated organs, and/or whole animals.
Binding versus Functional Assays
Binding assays used to be the method of choice for primary
pharmacological evaluation, mainly due to the ease of these assays compared to
functional assays which generally required more steps than binding assays.
However, several factors have changed this perception: (1) biotechnological
Functional assays have evolved profoundly and have decreased the number of assay
steps and increased the throughput dramatically, (2) functional assay equipment
has been automated, (3) ligand binding requires a high-affinity ligand, which
for many targets identified in genome projects simply does not exist, (4)
binding assays are generally unable to discriminate between agonists and
antagonists, (5) binding assays will generally only identify compounds binding
to the same site as the radioactively labeled tracer. One important aspect of
binding assays is the ability to determine ligand–receptor kinetics (on-rate,
off-rate, and ligand residence time) which are important pharmacological
properties affecting drug efficacy in vivo.
The Fluorometric Imaging Plate Reader (FLIPR™) illustrates this
development toward functional assays. Cells transfected with a receptor coupled
to increase in intracellular calcium levels (e.g., a Gαq-coupled
GPCR or a Ca2+ permeable ligand-gated ion channel) are loaded with the dye
Fluo-3 which in itself is not fluorescent. However, as shown in Figure, the dye
becomes fluorescent when exposed to Ca2+ in the cell in a
concentration-dependent manner. In this manner, ligand concentration–response
curves can be generated on the FLIPR very fast as it automatically reads all
wells of a 96-, 384-, or 1534-well tissue culture plate. Many other functional
assays along these lines have been developed in recent years. Importantly, the
majority of these assays can be applied on both recombinant and native receptor
expressing cell lines.
(a) Relation between Ca2+ concentration and relative fluorescence
intensity of the fluorescent probe fluo-3. (b) The 5-HT2B receptor subtype
belongs to the superfamily of G protein-coupled receptors and is coupled to
increase in inositol phosphates and intracellular Ca2+. Cells expressing 5-HT2B
receptors were loaded with fluo-3 and the fluorescence was determined upon
exposure to the endogenous agonist 5-HT (•)
and the partial agonists MK-212 (◦) and 2-Me-5-HT (◾)
on a FLIPR™.
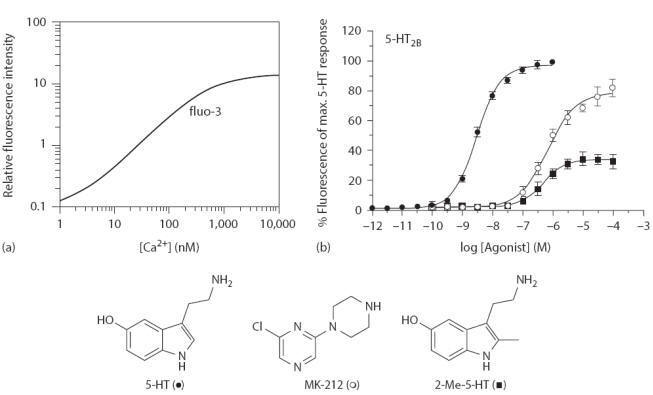
Partial and Full Agonists
Agonists are characterized by two pharmacological parameters:
potency and maximal response. The most common way of describing the potency is
by measurement of the agonist concentration which elicit 50% of the compound’s
own maximal response (the EC50 value). The maximal response is commonly
described as percent of the maximal response of the endogenous agonist. The
maximal response is also often described as efficacy or intrinsic activity which
were defined by Stephenson and Ariëns, respectively. Compounds, such as
2-Me-5-HT and MK-212 in Figure, show a lower maximal response than the
endogenous agonist and are termed partial agonists. The parameters potency and
maximal response are independent of each other and on the same receptor it is
thus possible to have, e.g., a highly potent partial agonist and a low potent
full agonist. Both parameters are important for drug research, and it is thus
desirable to have a pharmacological assay system which is able to determine both
the potency and the maximal response of the tested ligands.
Antagonists
Antagonists do not activate the receptors but block the activity
elicited by agonists and accordingly they are only characterized by the
parameter affinity. The most common way of characterizing antagonists is by
competition with an agonist (functional assay) or a radioactively labeled ligand
(binding assay). In both cases, the antagonist concentration is increased and
displaces the agonist or radioligand, which are held at a constant
concentration. It is then possible to determine the concentration of antagonist,
which inhibits the response/binding to 50% (the IC50 value). The IC50 value can
then be transformed to affinity (K) by the Cheng–Prusoff equation:
Functional assay:
K = IC50/(1 + [Agonist]/EC50)
(1)
where
[Agonist] is the agonist concentration
EC50 is for the agonist in the particular assay
Binding assay:
K = IC50/(1 + [Radioligand]/KD)
(2)
where
[Radioligand] is the radioligand concentration
KD is the affinity of the radioligand
It is important to observe that the Cheng–Prusoff equation is
only valid for competitive antagonists.
The Schild analysis is often used to determine whether an
antagonist is competitive or noncompetitive. In the Schild analysis, the
antagonist concentration is kept constant while the agonist concentration is
varied. For a competitive antagonist, this will cause a rightward parallel shift
of the concentration–response curves without a reduction of the maximal response
(Figure-a). The degree of right-shifting is determined as the dose ratio (DR),
which is the concentration of agonist giving a particular response in the
presence of antagonist divided by the concentration of agonist that gives the
same response in the absence of antagonist. Typically, one will chose the EC50
values to calculate the DR. In the Schild analysis, the log (DR-1) is depicted
as a function of the antagonist concentration (Figure-b). When the slope of the
curve equals 1, it is a sign of competitive antagonism and the affinity can then
be determined by the intercept of the abscissa. When the slope is significantly
different from 1 or the curve is not linear, it is a sign of noncompetitive
antagonism, which invalidates the Schild analysis.
Schild analysis of the competitive antagonist S16924 on cells
expressing the 5-HT2C receptor.
(a) Concentration–response curves of the agonist 5-HT were
generated in the presence of varying concentrations of S16924. Note the parallel
right shift of the curves and the same level of maximum response.
(b) Dose ratios are calculated and plotted as a function of the
constant antagonist concentration generating a straight line with a slope of
1.00
±
0.012. These results and the observations from (a) are in
agreement with a competitive interaction and the antagonist affinity can thus be
determined by the intercept of the abscissa;
K
= 12.9 nM.

As shown in the example in Figure, five concentration–response
curves are generated to obtain one antagonist affinity determination,
illustrating that the Schild analysis is rather work-intensive compared to,
e.g., the transformation by the Cheng–Prusoff equation where one inhibition
curve generates one antagonist affinity determination. However, the latter
cannot be used to determine whether an antagonist is competitive or
noncompetitive, which is the advantage of the Schild analysis.
When testing a series of structurally related antagonists one
would thus often determine the nature of antagonism with the Schild analysis for
a couple of representative compounds. If these are competitive antagonists, it
is reasonable to assume that all compounds in the series are competitive and
thus determine the affinity of these by the use of the less work-intensive
Cheng–Prusoff equation.
Constitutively Active Receptors and
Inverse Agonism
Most receptors display no or only minor basal activity but some
receptors display increased basal activity in the absence of agonist, which has
been referred to as constitutive activity. Interestingly, it has been shown that
inverse agonists can inhibit this elevated basal activity, which contrast
antagonists that inhibit agonist-induced responses but not the constitutive
activity (Figurea). Examples of important constitutively active receptors
include the human ghrelin receptor and several viral receptors that display
constitutive activity when expressed in the host cell. This latter group
includes the ORF-74 7TM receptor from human herpesvirus 8 (HHV-8), which show a
marked increased basal response when expressed in recombinant cells (Figure
12.13b). ORF-74 is homologous to chemokine receptors and does indeed bind
chemokine ligands. As shown in Figure 12.13b, chemokines display a wide range of
activities on the receptor from full agonism (e.g., GROα)
to full inverse agonism (e.g., IP10), which correlates with the
angiogenic/angiostatic effects of the chemokines. Constitutive activity can also
be caused by somatic mutations. Known examples include constitutively activating
mutations in the thyrotropin receptor and the luteinizing hormone receptor which
leads to adenomas, and the rhodopsin receptor which leads to night blindness.
(a) The nomenclature of ligand efficacies and schematic
illustration of their concentration dependent effects on constitutive activity.
(b) Ligand regulation of the constitutively active ORF-74 receptor from human
herpesvirus 8 (HHV8). ORF-74 is a G protein-coupled receptor coupled to
phosphatidylinositol (PI) turnover, which is regulated by a variety of human
chemokines ranging from full agonism by GROα
to full inverse agonism by IP10.
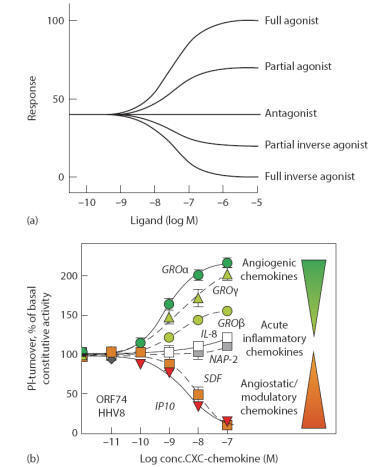
Allosteric Modulators
Allosteric modulators can both be stimulatory or inhibitory
(noncompetitive antagonists) and typically these compounds bind outside the
orthosteric binding site (Figure). Allosteric modulators have a number of
potential therapeutic benefits compared to agonists and competitive antagonists
which has led to significant increased pharmaceutical interest in recent years.
This increased interest has also been fueled by the development of functional
high-throughput screening assays which has made it possible to screen for
allosteric modulators.
The allosteric modulators mentioned below act through allosteric
mechanisms as evident from the fact that they do not displace radiolabeled
orthosteric ligands. Furthermore, their activity is dependent on the presence of
agonists as they do not activate the receptors by themselves. The fact that they
bind outside of the orthosteric ligand binding pocket often leads to increased
receptor subtype selectivity. Evolutionary pressure has led to conservation of
the orthosteric binding site at different subtypes, as radical mutations would
severely impact the binding properties. Thus, it is often seen that the
orthosteric binding site is much more conserved than the remaining part of the
receptor and accordingly, ligands binding to an allosteric site have a higher
chance of being selective. Likewise, the allosteric ligands will have a
different pharmacophore than the endogenous ligand which might improve, e.g.,
bioavailability. For example, ligands acting at the orthosteric site of the
GABAA receptor need a negatively charged acid function and a positively charged
basic function which greatly impairs the transport through biomembranes, whereas
allosteric ligands such as the benzodiazepine diazepam does not have any charged
groups and show excellent bioavailability. It is well known that many agonists,
particularly full agonists, lead to desensitization and internalization of
receptors (Fig). Unlike agonists, the positive modulators should prevent the
development of tolerance (as seen for, e.g., morphine), because they merely
potentiate the endogenous temporal receptor activation pattern and avoid
prolonged receptor activation leading to desensitization and internalization.
The fact that the receptors are stimulated in a more natural way by positive
modulators rather than the prolonged receptor activation caused by agonists may
also lead to a difference in physiological effects which may or may not be an
advantage.
Schild analysis of the noncompetitive antagonist fenobam on cells
expressing the metabotropic glutamate receptor subtype mGluR5.
Concentration–response curves of the agonist quisqualate were generated in the
presence of varying concentrations of fenobam. In contrast to the Schild
analysis shown in one before previous Figure, a clear depression of the maximal
response is seen with increasing antagonist concentrations. This shows that the
antagonist is noncompetitive. The localization of the orthosteric and allosteric
binding sites is depicted in previous Figure.
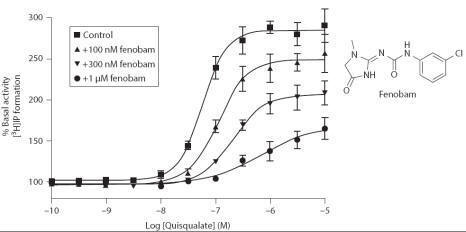
Negative Allosteric Modulators (Noncompetitive Antagonists)
As noted in the previous section, the Schild analysis is very
useful to discriminate between competitive and noncompetitive antagonists, and
an example of the latter is shown in Figure. Fenobam is a selective antagonist
at the mGluR5 receptor, and the Schild analysis clearly demonstrates that the
antagonism is noncompetitive due to the depression of the maximal response
(compare Figures). As noted previously, glutamate binds to the large
extracellular amino-terminal domain whereas fenobam has been shown to bind to
the extracellular part of the 7TM domain. Fenobam does not hinder binding of
glutamate to the extracellular domain, but hinder the conformational change
leading to receptor activation.
Positive Allosteric Modulators
Positive allosteric modulation can be achieved through several
mechanisms. For example, benzodiazepines positively modulate the GABAA receptor
by increasing the frequency of channel opening. Positive modulation can also be
obtained by blocking receptor desensitization as exemplified by cyclothiazide.