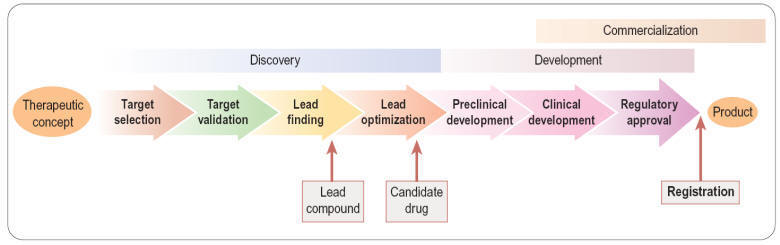
Figure - Three main
phases of the creation of a new drug: discovery, development and
commercialization.
DRUG DISCOVERY PROCESS
Process Overview
Target Identification and Validation
The process of drug discovery
starts long before the screening of compound libraries for molecules that bind
to and affect the action of a protein associated with disease. Drug discovery
starts with the identification of the disease associated protein or molecular
target. In the past, identification of molecular targets was a painstakingly
slow process, generally carried out by investigators who could spend entire
careers studying one pathway or one protein that is involved in some aspect of
human development, metabolism, or disease. In the course of defining the role of
a given protein in the cellular life cycle, the direct relevance of this protein
to a disease state would often become clear. By removing or altering the
target’s function from the cell, the target would then be validated as a
suitable target for drug screening. In recent times, methods to identify panels
of targets that are associated with particular pathways or disease states have
emerged, and the acceleration of target identification has led to a bottleneck
in target validation. Methods described in this chapter are being deployed to
reduce the bottleneck and increase the availability of new, useful,
disease-associated molecular targets for development of new classes of effective
therapeutic compounds.
Screening for Hits
Once a disease-associated molecular target has been identified and validated in
disease models, screening for a selective and potent inhibitor (or activator) of
the target is the next step. Libraries of compounds that are either synthetic
chemicals, peptides, natural or engineered proteins, or antibodies are exposed
to the target in a manner that will detect and isolate those members of the
library that interact with and, preferably, have an effect on the target. The
compounds selected are called hits. Initially, screening can be performed
by searching for compounds that bind to the target, but binding is not
sufficient for therapeutic activity. More recent screening procedures include an
activity-based readout as part of the initial screening assay. For example, if
the goal is to inhibit a protein that is involved in activating the expression
of a particular gene or set of genes, the assay can include a readout to
determine if the expression of the gene is reduced by the compound. Such assays
can be cell-based, but more often they are enzymatic assays that can be
performed in a high-throughput manner.
Lead Optimization
Once the initial screening is performed, a large collection of hits is obtained.
These hits are then evaluated and the best ones identified in a process known as
lead optimization. During this stage, scientists determine which, if any,
of the hits selected has the appropriate properties to justify continued
expenditure of resources on the development of the compound as a clinical
candidate. These properties include ease of synthesis; adherence to the Lipinsky
rules, which describe chemical characteristics that are predictive of
biodistribution and in vivo activity; specificity for the target; and efficacy
in the disease. The hits discovered during the screening process are therefore
characterized by a variety of biochemical, biophysical, and biological methods
to narrow the set down to a handful of compounds. Assays that measure each
compound’s activity directly are a useful first step at determining potency and
can enable the most effective compounds to be identified. The resulting smaller
set of hits, or leads, is then tested in more rigorous models of disease,
including either cell-based or animal models of the
disease.
Pharmacology and Toxicology
Once one or more potential lead compounds emerge from the lead optimization
efforts, the compound must be evaluated in multiple disease models, and if
possible, compared to existing therapies for the same disease. Further,
properties of the compound are studied when delivered into a living organism.
The classic set of properties that must be appropriate include Absorption
(through the intended route of administration), distribution (what organs does
it end up in?), metabolism (what are the by-products of cellular metabolism of
the compound, and what potential effects do these compounds have on the organism
and disease?), and excretion (how is it processed and eliminated from the
body?). Collectively known as ADME, this set of properties is essential to
evaluate in multiple species, in addition to measuring efficacy in sophisticated
disease models. Design of ADME and efficacy experiments is a crucial activity in
drug discovery and development. All drugs that are intended for human clinical
applications must be tested for toxicity using the same material that will be
used in humans, manufactured by the same process. Toxicology testing is a highly
regulated process that is governed by regulatory authorities such as the U.S.
Food and Drug Administration (FDA). Developing a manufacturing process for a
drug (which is outside the scope of this chapter) and executing the necessary
toxicology program are very expensive and are therefore usually performed on
only one lead compound identified during lead optimization and subsequent
efficacy testing. Only if the compound fails at this stage will another compound
from the same screening (if possible) be advanced into preclinical development.
Different companies, and in fact countries, have different standards for which
products move into human clinical testing, but in all cases the product’s safety
must be adequately demonstrated before regulatory approval can be obtained for
advancing the product into humans.
Clinical Trials
Human clinical testing follows an established process in most regulated
countries worldwide. If possible, the initial administration of a compound to a
human subject is performed on people who do not have the disease indication for
which the product is designed. The compound is administered at a dose that is
significantly lower than the intended therapeutic dose, and the trial subjects
are monitored for any signs of toxicity. Most often, these adverse events or
side effects consist of headache, fever, nausea, or other discomforts. If the
trial subjects receiving the initial low dose have no or minimal side effects,
the next cohort of subjects is treated with a higher dose. Using this dose
escalation method, the aim is to determine the maximum tolerated dose (MTD) or
to determine the safety of a dose level that is known to generate therapeutic
benefit. Depending on the disease indication, the tolerance for adverse
reactions is different. For example, almost all chemotherapeutic agents for
cancer are significantly toxic, but many cancer patients are willing to suffer
these effects in order to have a chance to overcome their disease and continue
to live without cancer. Due to their known toxicity, oncology drugs are
therefore rarely tested in normal volunteers, but in fact are often tested
initially in patients who have failed all other chemotherapy or other treatment
regimens and have no other option for survival. Chronic, nonfatal diseases,
however, are not usually treated with agents that have severe side effects
because the benefit of the drug does not outweigh the risks and discomfort of
the side effects. Once the MTD has been determined, drugs usually move into
phase II testing, in which the drug’s efficacy is determined in a small select
group of patients who have the disease for which the drug was developed. These
trials can also include a range of doses and dosing regimens (modes of delivery,
frequency, etc.), in order to measure the clinical efficacy of different doses.
Designing phase II trials and choosing endpoints or objectives for the therapy
that will accurately reflect the compound’s efficacy is a very demanding
process, and many compounds can fail at this stage. Only 60% of drugs that enter
phase II successfully complete this stage and move on to the pivotal phase III
trial.
Phase III is considered
pivotal because the drug is administered to a much larger group of patients and
is evaluated for efficacy with greater rigor than in phase II. These trials can
cost millions of dollars and therefore are attempted only when the phase II
results are highly convincing. There are regulatory hurdles that must be crossed
to enter phase III. The drug product that is used in phase III must be
manufactured exactly as the first commercial product will be made. The facility
or reactor that is used, the process, the analytical methods, and the
formulation and vialing are all identical to the intended final product that
will be made and launched upon product approval. Following the successful
completion of phase III clinical testing, the owner of the compound then fi les
an application with regulatory authorities in various countries, such as the
U.S. FDA, for permission to sell the compound for the intended therapeutic
indication. The FDA or other agency must then grant a license to market the
compound, and the new drug is launched onto the market.
Motivation for Improvement
The drug discovery and
development process described above follows a logical and linear path from
target identification through completion of clinical trial and submission of an
application to the regulatory authorities seeking approval to market a drug.
However, this process is lengthy, cumbersome, and most important, generates
compounds that are more likely than not to fail during clinical testing. The
major hurdles in drug discovery and development today are:
• Time. From the
initial discovery and validation of a molecular target to final marketing of an
effective drug can take 10 to 15 years.
• Efficiency. a
significant portion of the high cost of drug development is due to the very high
attrition rate of compounds that enter the clinic; only 12% succeed.
• Expense. Drug
development can cost up to $800 million per successful compound.
Therefore, improved
clinical outcomes of therapeutic candidates at all stages of development would
contribute significantly to reducing development costs and improving the number
of candidates that succeed in obtaining approval for commercialization. New
methods that enable better target identification and validation, a deeper
understanding of total systems biology and the implications for any specific
drug or target in development, and methods to understand the nature of molecular
targets to enable more effective screening and lead identification would be
extremely valuable in meeting the needs for cost-effective development of new
and useful therapeutics for human disease. The emerging technologies described
in this chapter aim to address the many aspects of drug development and to
enable more effective understanding of targets and compounds through
high-throughput high-content analysis.

Target Identification
The initial stage of the
drug discovery process is the identification of a molecular target, almost
always a protein, which can be shown to be associated with disease. To
accelerate this, more proteins need to be identified and characterized. Although
the set of human genes is by now quite well understood, this is not as true for
all the proteins in the human proteome. Because of splice variants and
posttranslational modifications, the number of proteins far exceeds the number
of genes. As of today, a great many human proteins have yet to be fully
identified and characterized. The new high-throughput technologies of proteomics
are helping to close this knowledge gap in a number of ways:
1. Identify large
numbers of novel proteins in the search for new drug targets. High
throughput separation, purification, and identification methods are being used
for this. 2DPAGE is often used for separation, even though it has some
drawbacks. HPLC is a common separation and purification technique, and is
particularly adapted for high-throughput use when coupled directly to tandem MS
for identification. These procedures produce peptide sequences that must then be
compared to possible homologs in other organisms (or paralogs in humans) using
informatics. Edman sequencing may be needed for sequence verification.
2. Perform initial
functional characterization of novel proteins. Initially, assign them to
functional areas that might implicate some of them in disease pathology:
a. Metabolic pathways
b. Transcriptional
regulatory networks
c. Cell cycle regulatory
networks
d. Signal transduction
pathways
Computational sequence
homology analysis can help putatively assign a novel protein to a known pathway
or network that its homolog participates in. Sequence analysis also helps
identify known motifs and functional domains that the new protein shares with
known proteins. This information can provisionally assign membership in a
protein family to the new protein.
3. Screen a compound
with known therapeutic effect against a large number of human proteins to
identify the exact target. It is not uncommon to have a compound with known
but perhaps limited or suboptimal therapeutic effect against a disease, yet not
know the molecular target for this compound. A proteome-scale search can
pinpoint a possible target, once binding has been identified. The case study of
the development of LAF389, presented at the end of this section, describes a
successful example of this in detail.
4. Perform
high-throughput differential protein expression profiling, comparing diseased
with normal tissue samples, to identify the biomarker proteins that are possible
contributors to the disease by their over- or underexpression. This is one
of the most promising areas of application of proteomics in target
identification. Several proteomics technologies can be used for expression
profiling. Two-dimensional PAGE has been used, although comparing two different
gels (one for normal, one for disease state) can be problematic, due to the
replicability issue discussed earlier. A variant of this called
two-dimensional differential in-gel electrophoresis (2D-DIGE) overcomes this
drawback. 2D-DIGE has been used successfully to identify biomarkers that could
be potential drug targets. As noted previously, protein chips have perhaps the
greatest potential for high-throughput simultaneous differential protein
expression profiling. Several successful attempts to use protein arrays for
target identification have been reported. However, the technology is still
relatively immature and is therefore further out on the horizon as far as
general industry acceptance for target identification.
Example
A
recent study showed how an integrated approach involving proteomics,
bioinformatics, and molecular imaging was used to identify and characterize
disease-tissue specific signature proteins displayed by endothelial cells in the
organism. Working with blood vessels in normal lungs and in lung tumors in rats,
the researchers used several high-throughput affinity-based separation
procedures, followed by MS and database analysis to identify and map the
proteins displayed by endothelial cells that line the blood vessels. With this
approach they identified proteins that are displayed only on solid tumor blood
vessel walls. They then demonstrated that radioisotope-labeled antibodies can
recognize these tumor-specific proteins, allowing them to be imaged. The
radioisotope labeling itself also resulted in significant remission of solid
lung tumors, demonstrating tissue-targeted therapeutic potential. Although
individual endothelial proteins had been identified and targeted in previous
studies, this is the first time a proteomics approach had been used to move
toward a complete tissue-specific mapping of the proteins displayed on the
blood-exposed surfaces of blood vessels. This demonstrates a new approach for
identifying potential novel targets for therapy.
Target Validation
At this point in the drug
discovery process, one or more potentially disease-related protein targets have
been identified. For now, let us assume one. The next step is to validate the
target. Primarily, this means that the target’s relevance to disease pathology
must be determined unambiguously. This involves more detailed functional
characterization, more evidence for the pathway or network assignment, and
modulating the protein’s activity to determine its relationship to the disease
phenotype. Some determination of tractability may be done in this stage as well.
Proteomics can assist in several ways:
1. Determine in what
tissues and cell components it appears and in which developmental stages.
High-throughput techniques such as protein chips and 2D-DIGE can be used for
proteome-scale expression studies comparing different tissue types and
developmental stages. These add evidence that the putative target is found in a
disease-related tissue and at the expected developmental stage. Interaction
studies can help determine subcellular localization by showing binding to
proteins or phospholipids in the cell that have known location. Further,
sequence analysis can identify known location-specifying signal peptides on the
protein. Finally, posttranslational modification analysis can identify certain
PTMs that determine the destination of the protein.
2. Understand when and
for how long the target gets expressed, and when degraded.
High-throughput protein
expression studies can be done in multiple runs over time, and then compared. In
this case, expression patterns are assayed and then compared across multiple
time points rather than normal vs. disease states. For additional evidence it
would be very informative to do a time-based expression study first of normal
tissue, then another one of diseased tissue, and compare the behavior over
time of normal vs. disease tissues. This would provide a multidimensional
target validation.
3. Verify the target
protein’s specific role within the protein family and the pathway
or
network identified in the previous stage.
Initial putative functional
assignments for the new protein were made in the target identification stage. In
target validation, high throughput protein–protein interaction studies can be
used to strengthen the evidence for the protein family, network, or pathway
involvement. Protein–phospholipid interaction assays can determine whether the
new protein is membrane associated. Technologies such as protein chips are
beginning to be used for these interaction studies and have great potential in
this area. Other techniques such as Y2H, phage display, and tandem affinity
purification have also been used with success in this area.
Posttranslational
modifications to the new protein can also be identified by methods discussed
earlier. Knowledge of the binding partners and posttranslational modifications
of a new protein goes a long way to help characterize it functionally, to
solidify its assignment to a pathway, and so on. Since the association of a
disease and a particular function or pathway is often already known, a solid
assignment of the new protein to such a function or pathway implicates the
protein in the disease. This adds evidence toward validating the target.
4. Determine the effect
of inhibiting the putative target.
a. Does target inhibition
disrupt a disease-related pathway?
b. Does target inhibition
slow or stop uncontrolled growth? Have other effects?
c. Does this effect
validate the target as unambiguously related to the disease pathology?
Several methods can be
used to answer these questions. Gene knockout studies in mice have been an
effective tool for some time, but knockouts cannot be done with all putative
target proteins. It is in any case a slow, laborious process to breed the
knockout strain correctly and reliably and may not result in viable mice to
study.
One recent alternative
method has garnered a great deal of attention and is currently achieving rapid
adoption in the industry due to its relative ease, rapidity, effectiveness, and
lower cost. It is called RNA interference (RNAi). This technique has
roots in both genomics and proteomics. Small interfering RNAs (siRNA) are
synthetic 19- to 23-nucleotide RNA strands that fold in hairpin configuration.
They elicit strong and specific suppression of gene expression; this is called
gene knockdown or gene silencing. RNAi works by triggering the
degradation of the mRNA transcript for the target’s gene before the protein can
be formed. RNAi is done in vitro in the lab to verify disease relevance of a
putative target in several possible ways:
• If the target is
believed to cause pathology by overexpression, investigators can knock
down the gene for the target and observe whether the disease pathology is
reduced.
• If underexpression
is assumed to contribute to disease, the gene can be knocked down in
healthy tissue samples to see whether this elicits the same disease
phenotype.
• The pathway in which the
target is believed to participate can itself be validated as disease-related by
performing knockdowns of some or all of the genes in the pathway.
Then the putative target
gene can be knocked down, and the effect of this on the pathway’s function can
be observed. This both validates disease relevance and verifies the functional
assignment of the target to the pathway. Note that RNAi is also being
investigated for its therapeutic potential. Although there are major ADME
issues to be addressed, RNAi molecules, with their high specificity and efficacy
in gene suppression, may themselves hold great promise as drug candidates.
Screening for Hits
Now we have one or more
possible targets. For each target we need to screen many compounds to look for
drug candidates that show activity against the target (i.e., hits). This can be
like searching for a needle in a haystack, so any techniques that can help
accelerate and focus this search are of great value. The following techniques
are fairly new but are in current use.
1. Develop sets of
compounds to screen for activity against the target. The first order of
business is to construct focused sets of compounds to screen against the target.
Structural proteomics and combinatorial chemistry can play major roles at this
stage. As mentioned before, an x-ray crystallographic structure for the protein
provides the golden standard for three-dimensional structure. Given such
structural information, it is possible to develop much more focused compound
sets for screening libraries than would otherwise be possible. Combinatorial
chemistry can then be used to design the libraries of such compounds. These are
numerous small modifications of a basic small molecule or side group that is
likely to fit the known binding pocket of the target protein based on the
structural information. Computational chemistry is also adding to this
effort. It is being used to generate large virtual compound libraries as
a part of structure-based drug design. Like the combinatorial compound
libraries, these are sets of compounds that are likely to fit well with the
target’s binding site. However, these compounds are all in silico, in the
computer only.
2. Screen for compounds
that affect the target. (In most cases, inhibition is aimed for. Most drug
targets are enzymes, and most of these are overactive, either by being
overabundant or by being stuck in an active state. Therefore, most drug
candidates attempt to inhibit or shut off the enzyme by binding to its active
site.)
High-throughput laboratory
screening can now proceed. This technique uses 96- or 384-well plates to combine
the target protein with each of the screening compounds, one per well. In this
way, the compounds are individually tested for activity against the target. Once
activity is detected, even if it is only moderate, that compound is designated
as a hit. Chemists can later attempt to increase its activity by modifying the
compound and retesting.
Virtual screening
is
another, newer technique that is showing some promise. Using the virtual
compound libraries, virtual screening uses elaborate computational chemistry
techniques to determine in silico the fi t between each virtual compound
model and the binding site of the protein model derived from the x-ray
crystallographic structure. This involves computing the chemical affinity, the
steric fi t, and the energetics of the compound in the binding pocket. When
certain thresholds are reached, a hit is declared, and reserved for further
optimization.
3. Structure-based drug
design. As mentioned above, virtual screening entails the computational
identification of a drug candidate from the ground up. This is called
structure based or rational drug design. Structural proteomics
provides the core information needed to achieve this. There are two main
approaches used in structure-based drug design:
a. Building up an
optimized ligand from a known inhibitor molecule. If structural information
is available for a known inhibitor ligand, this can be modeled and used as a
starting point. Placed computationally into the target protein’s binding site,
it can be manipulated on the computer by chemical changes or the addition,
moving, or subtraction of chemical groups or even atoms until its fi t is
considered strong. Some docking programs first decompose the known ligand into
fragments before the user docks them appropriately in the binding pocket and
begins optimizing.
b. De novo ligand
design and/or docking into the binding pocket model. If no known inhibitor
exists, or there is no structural information on one, a ligand can be built up
from scratch. A base fragment is initially placed in the binding site, then
additional fragments or atoms are added according to sets of rules derived from
many known
protein–ligand structures.
Biochemical and cell-based assays
There is a wide range of assays formats that can be deployed in
the drug discovery arena, although they broadly fall into two categories:
biochemical and cell-based.
Biochemical assays (Figure) involve the use of cell-free in-vitro systems to model
the biochemistry of a subset of cellular processes. The assay systems vary from
simple interactions, such as enzyme/substrate reactions, receptor binding or
protein–protein interactions, to more complex models such as in-vitro
transcription systems. In contrast to cell-based assays, biochemical assays give
direct information regarding the nature of the molecular interaction (e.g.
kinetic data) and tend to have increased solvent tolerance compared to cellular
assays, thereby permitting the use of higher compound screening concentration if
required. However, biochemical assays lack the cellular context, and are
insensitive to properties such as membrane permeability, which determine the
effects of compounds on intact cells.

Figure - Types of biochemical
assay.
Unlike
biochemical assays, cell-based assays (Figure) mimic
more closely the in-vivo situation and can be adapted for targets that
are unsuitable for screening in biochemical assays, such as those involving
signal transduction pathways, membrane transport, cell division, cytotoxicity
or antibacterial actions. Parameters measured in cell-based assays range from
growth, transcriptional activity, changes in cell metabolism or morphology, to
changes in the level of an intracellular messenger such as cAMP, intracellular
calcium concentration and changes in membrane potential for ion channels.
Importantly, cell-based assays are able to distinguish between receptor
antagonists, agonists, inverse agonists and allosteric modulators which cannot
be done by measuring binding affinity in a biochemical assay.
Many
cell-based assays have quite complex protocols, for example removing cell
culture media, washing cells, adding compounds to be tested, prolonged
incubation at 37°C, and, finally, reading the cellular response. Therefore,
screening with cell-based assays requires a sophisticated infrastructure in the
screening laboratory (including cell cultivation facilities, and robotic systems
equipped to maintain physiological conditions during the assay procedure) and
the throughput is generally lower.
Cell-based
assays frequently lead to higher hit rates, because of non-specific and
‘off-target’ effects of test compounds that affect the readout. Primary hits
therefore need to be assessed by means of secondary assays such as non- or
control-transfected cells in order to determine the mechanism of the effect.
Although cell-based assays are generally more time-consuming than cell-free
assays to set up and run in high-throughput mode, there are many situations in
which they are needed. For example, assays involving G-protein coupled receptors
(GPCRs), membrane transporters and ion channels generally require intact cells
if the functionality of the test compound is to be understood, or at least
membranes prepared from intact cells for determining compound binding. In other
cases, the production of biochemical targets such as enzymes in sufficient
quantities for screening may be difficult or costly compared to cell-based
assays directed at the same targets. The main pros and cons of cell-based assays
are summarized in
Table.

Figure - Types of cell-based
assay.
Radiological
Ligand binding assays
Assays to
determine direct interaction of the test compound with the target of interest
through the use of radiolabelled compounds are sensitive and robust and are
widely used for ligand-binding assays. The assay is based on measuring the
ability of the test compound to inhibit the binding of a radiolabelled ligand to
the target, and requires that the assay can distinguish between bound and free
forms of the radioligand. This can be done by physical separation of bound from
unbound ligand (heterogeneous format) by filtration, adsorption or
centrifugation. The need for several washing steps makes it unsuitable for fully
automated HTS, and generates large volumes of radioactive waste, raising safety
and cost concerns over storage and disposal. Such assays are mainly restricted
to 96-well format due to limitations of available multiwell filter plates and
achieving consistent filtration when using higher density formats. Filtration
systems do provide the advantage that they allow accurate determination of
maximal binding levels and ligand affinities at sufficient throughput for
support of hit-to-lead and lead optimization activities. In the HTS arena,
filtration assays have been superseded by homogeneous formats for
radioactive assays. These have reduced overall reaction volume and eliminate the
need for separation steps, largely eliminating the problem of waste disposal and
provide increased throughput.

The
majority of homogenous radioactive assay types are based on the scintillation
proximity principle. This relies on the excitation of a scintillant incorporated
in a matrix, in the form of either microbeads (’SPA’) or microplates
(Flashplates™, Perkin Elmer Life and Analytical Sciences), to the surface of
which the target molecule is also attached (Figure). Binding of the radioligand to the target brings it into close
proximity to the scintillant, resulting in light emission, which can be
quantified. Free radioactive ligand is too distant from the scintillant and no
excitation takes place. Isotopes such as 3H or 125I are typically used, as they
produce low-energy particles that are absorbed over short distances. Test
compounds that bind to the target compete with the radioligand, and thus reduce
the signal.
With bead technology, polymer beads of ~5 μm diameter are coated
with antibodies, streptavidin, receptors or enzymes to which the radioligand can
bind. Ninety-six- or 384-well plates can be used. The emission wavelength of the
scintillant is in the range of 420 nm and is subject to limitations in the
sensitivity due to both colour quench by yellow test compounds, and the variable
efficiency of scintillation counting, due to sedimentation of the beads. The
homogeneous platforms are also still subject to limitations in throughput
associated with the detection technology via multiple photomultiplier tube-based
detection instruments, with a 384-well plate taking in the order of 15 minutes
to read.
The drive for increased throughput for radioactive assays led to
development of scinitillants, containing europium yttrium oxide or europium
polystyrene, contained in beads or multiwell plates with an emission wavlength
shifted towards the red end of the visible light spectrum (~560 nm) and suited
to detection on charge-coupled device (CCD) cameras. The two most widely adopted
instruments in this area are LEADseeker™ (GE Healthcare) and Viewlux™ (Perkin
Elmer), using quantitative imaging to scan the whole plate, resulting in a
higher throughput and increased sensitivity. Imaging instruments provide a read
time typically in the order of a few minutes or less for the whole plate
irrespective of density, representing a significant improvement in throughput,
along with increased sensitivity. The problem of compound colour quench effect
remains, although blue compounds now provide false hits rather than yellow. As
CCD detection is independent of plate density, the use of imaging based
radioactive assays has been adopted widely in HTS and adapted to 1536-well
format and higher.
In the microplate form of scintillation proximity assays the
target protein (e.g. an antibody or receptor) is coated on to the floor of a
plate well to which the radioligand and test compounds are added. The bound
radioligand causes a microplate surface scintillation effect. FlashPlate™ has
been used in the investigation of protein–protein (e.g. radioimmunoassay) and
receptor–ligand (i.e. radioreceptor assay) interactions, and in enzymatic (e.g.
kinase) assays.
Due to
the level of sensitivity provided by radioactive assays they are still widely
adopted within the HTS setting. However, environmental, safety and local
legislative considerations have led to the necessary development of alternative
formats, in particular those utilizing fluorescent-ligands. Through careful
placement of a suitable fluorophore in the ligand via a suitable linker, the
advantages of radioligand binding assays in terms of sensitivity can be realized
without the obvious drawbacks associated with the use of radioisotopes. The use
of fluorescence-based technologies is discussed in more detail in the following
section.
Fluorescence technologies
The
application of fluorescence technologies is widespread, covering multiple
formats and yet in the simplest form involves excitation of a sample with light
at one wavelength and measurement of the emission at a different wavelength. The
difference between the absorbed wavelength and the emitted wavelength is called
the Stokes shift, the magnitude of which depends on how much energy is
lost in the fluorescence process. A large Stokes shift is advantageous as it
reduces optical crosstalk between photons from the excitation light and emitted
photons.
Fluorescence techniques currently applied for HTS can be grouped
into six major categories:
•
Fluorescence intensity
•
Fluorescence resonance energy transfer
•
Time-resolved fluorescence
•
Fluorescence polarization
•
Fluorescence correlation
•
AlphaScreen™ (amplified luminescence proximity homogeneous
assay).
Fluorescence intensity
In
fluorescence intensity assays, the change of total light output is monitored and
used to quantify a biochemical reaction or binding event. This type of readout
is frequently used in enzymatic assays (e.g. proteases, lipases). There are two
variants: fluorogenic assays and fluorescence quench assays. In
the former type the reactants are not fluorescent, but the reaction products
are, and their formation can be monitored by an increase in fluorescence
intensity.
In fluorescence quench assays a fluorescent group is covalently
linked to a substrate. In this state, its fluorescence is quenched. Upon
cleavage, the fluorescent group is released, producing an increase in
fluorescence intensity.
Fluorescence intensity measurements are easy to run and cheap.
However, they are sensitive to fluorescent interference resulting from the
colour of test compounds, organic fluorophores in assay buffers and even
fluorescence of the microplate itself.
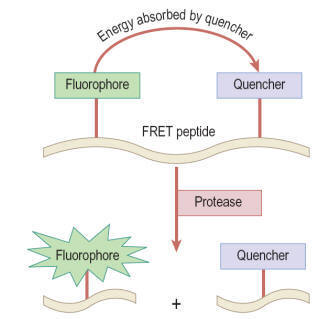
Fluorescence resonance energy transfer (FRET)
In this type of assay a donor fluorophore is excited and
most of the energy is transferred to an acceptor fluorophore or a
quenching group; this results in measurable photon emission by the acceptor. In
simple terms, the amount of energy transfer from donor to acceptor depends on
the fluorescent lifetime of the donor, the spatial distance between donor and
acceptor (10–100 Å), and the dipole orientation between donor and acceptor. The
transfer efficiency for a given pair of fluorophores can be calculated using
the equation of Förster.
Usually the emission wavelengths of donor and acceptor are
different, and FRET can be determined either by the quenching of the donor
fluorescence by the acceptor (as shown in
Figure)
or by the fluorescence of the acceptor itself. Typical applications are for
protease assays based on quenching of the uncleaved substrate, although FRET has
also been applied for detecting changes in membrane potential in cell-based
assays for ion channels. With simple FRET techniques interference from
background fluorescence is often a problem, which is largely overcome by the use
of time-resolved fluorescence techniques, described below.
Time resolved fluorescence (TRF)
TRF
techniques use lanthanide chelates (samarium, europium, terbium and dysprosium)
that give an intense and long-lived fluorescence emission (>1000 μs).
Fluorescence emission is elicited by a pulse of excitation light and measured
after the end of the pulse, by which time short-lived fluorescence has subsided.
This makes it possible to eliminate short-lived autofluorescence and reagent
background, and thereby enhance the signal-to-noise ratio. Lanthanides emit
fluorescence with a large Stokes shift when they coordinate to specific ligands.
Typically,
the complexes are excited by UV light, and emit light of wavelength longer than
500 nm. Europium (Eu3+) chelates have been used in immunoassays by means of a
technology called DELFIA (dissociation-enhanced lanthanide fluoroimmuno assay).
DELFIA is a heterogeneous time-resolved fluorometric assay based on
dissociative fluorescence enhancement. Cell-and membrane-based assays are
particularly well suited to the DELFIA system because of its broad detection
range and extremely high sensitivity. High sensitivity – to a limit of about
10−17 moles/well – is achieved by applying the dissociative enhancement
principle. After separation of the bound from the free label, a reagent is added
to the bound label which causes the weakly fluorescent lanthanide chelate to
dissociate and form a new highly fluorescent chelate inside a protective
micelle. Though robust and very sensitive, DELFIA assays are not ideal for HTS,
as the process involves several binding, incubation and washing steps.
The need for
homogeneous (‘mix and measure’) assays led to the development of LANCETM (Perkin
Elmer Life Sciences) and HTRF® (Homogeneous Time-Resolved Fluorescence;
Cisbio). LANCETM, like DELFIA®, is based on chelates of lanthanide ions, but in
a homogeneous format. The chelates used in LANCETM can be measured directly
without the need for a dissociation step, however in an aqueous environment the
complexed ion can spontaneously dissociate and increase background
fluorescence.
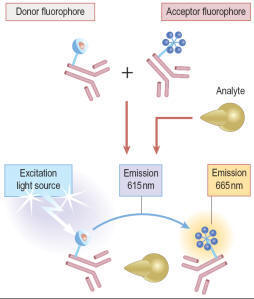
In HTRF® (Figure) these limitations are overcome by the use of a cryptate
molecule, which has a cage-like structure, to protect the central ion (e.g.
Eu+) from dissociation. HTRF® uses two separate labels, the donor (Eu)K and the
acceptor APC/XL665 (a modified allophycocyanine from red algae) and such assays
can be adapted for use in plates up to 1536-well format. In both LANCETM and
HTRF®, measurement of the ratio of donor and acceptor fluorophore emission can
be applied to compensate for non-specific quenching of assay reagents. As a
result, the applications of both technologies are widespread, covering detection
of kinase enzyme activity , protease activity, second messengers such as cAMP
and inositiol tri-phosphate (InsP3) and numerous biomarkers such as interleukin
1β (IL-1β) and tumour necrosis factor alpha (TNFα).
Fluorescence polarization (FP)
When a stationary molecule is excited with plane-polarized light
it will fluoresce in the same plane. If it is tumbling rapidly, in free
solution, so that it changes its orientation between excitation and emission,
the emission signal will be depolarized. Binding to a larger molecule reduces
the mobility of the fluorophore so that the emission signal remains polarized,
and so the ratio of polarized to depolarized emission can be used to determine
the extent of binding of a labelled ligand. The rotational relaxation speed
depends on the size of the molecule, the ambient temperature and the viscosity
of the solvent, which usually remain constant during an assay. The method
requires a significant difference in size between labelled ligand and target,
which is a major restriction to its application and the reliance on a single,
non-time resolved fluorescence output makes the choice of fluorphore important
to minimize compound interference effects. FP-based assays can be used in
96-well up to 1536-well formats.
Fluorescence correlation methods
Although an uncommon technique in most HTS departments, due the
requirement for specific and dedicated instrumentation, this group of
fluorescence technologies provide highly sensitive metrics using very low levels
of detection reagents and are very amendable to ultra-high throughput screening
(uHTS). The most widely applied readout technology, fluorescence correlation
spectroscopy, allows molecular interactions to be studied at the
single-molecule level in real time. Other proprietary technologies such as
fluorescence intensity distribution analysis (FIDA and 2-dimensional FIDA also
fall into this grouping, sharing the common theme of the analysis of
biomolecules at extremely low concentrations. In contrast to other fluorescence
techniques, the parameter of interest is not the emission intensity itself, but
rather intensity fluctuations. By confining measurements to a very small
detection volume (achieved by the use of confocal optics) and low reagent
concentrations, the number of molecules monitored is kept small and the
statistical fluctuations of the number contributing to the fluorescence signal
at any instant become measurable. Analysis of the frequency components of such
fluctuations can be used to obtain information about the kinetics of binding
reactions.
With help of the confocal microscopy technique and laser
technologies, it has become possible to measure molecular interactions at the
single molecule level. Single molecule detection (SMD) technologies provide a
number of advantages: significant reduction of signal-to-noise ratio, high
sensitivity and time-resolution. Furthermore, they enable the simultaneous
readout of various fluorescence parameters at the molecular level. SMD readouts
include fluorescence intensity, translational diffusion (fluorescence
correlation spectroscopy, FCS), rotational motion (fluorescence polarization),
fluorescence resonance energy transfer, and time-resolved fluorescence. SMD
technologies are ideal for miniaturization and have become amenable to
automation. Further advantages include very low reagent consumption and broad
applicability to a variety of biochemical and cell-based assays.
Single molecular events are analysed by means of confocal optics
with a detection volume of approximately 1 fL, allowing miniaturization of HTS
assays to 1 μL or below. The probability is that, at any given time, the
detection volume will have a finite number of molecular events (movement,
intensity, change in anisotropy), which can be measured and computed. The
signal-to-noise ratio typically achieved by these methods is high, while
interference from scattered laser light and background fluorescence are largely
eliminated.
Fluorescence lifetime analysis is a relatively straightforward
assay methodology that overcomes many of the potential compound intereference
effects achieved through the use of TRF, but without the requirement for
expensive fluorophores. The technique utilizes the intrinsic lifetime of a
fluorophore, corresponding to the time the molecule spends in the excited
state. This time is altered upon binding of the fluorophore to a compound or
protein and can be measured to develop robust assays that are liable to minimum
compound intereference using appropriate detection instrumentation.
AlphaScreen™ Technology
The proprietary bead-based technology from Perkin Elmer is a
proximity-based format utilizing a donor bead which, when excited by light at a
wavelength of 680 nm, releases singlet oxygen that is absorbed by an acceptor
bead, and assuming it is in sufficiently close proximity (<200 nm) this results
in the emission of light between 520 and 620 nm (Figure).
This phenomenon is unusual in that the wavlength of the emitted light is shorter
and therefore has higher energy than the excitation wavelength. This is of
significance since it reduces the potential for compound inner filter effects;
however, reactive functionality may still inhibit the energy transfer.

As with
other bead-based technologies the donor and acceptor beads are available with a
range of surface treatments to enable the immobilization or capture of a range
of analytes. The range of immobolization formats and the distance over which the
singlet oxygen can pass to excite the donor bead provide a suitable format for
developing homogeneous antibody-based assays similar to enzyme-linked
immunosorbent assays (ELISA) which are generally avoided in the HTS setting due
to multiple wash, addition and incubation steps. These bead-based ELISA, such as
AlphaLISA™ (Perkin Elmer), provide the required sensitivity for detection of
biomarkers in low concentration and can be configured to low volume 384-well
format without loss of signal window.
AlphaLISA
AlphaLISA is a further development of the
AlphaScreen technology that relies on the same donor beads but uses a different
type of acceptor beads. In AlphaLISA beads, anthracene and rubrene are
substituted by europium chelates. Excited europium emits light at 615 nm with a
much narrower wavelength bandwidth than AlphaScreen (fig.). Hence, AlphaLISA
emission is less susceptible to compound interference and can be employed for
the detection of analytes in biological samples like cell culture supernatants,
cell lysates, serum, and plasma.
AlphaLISA allows the
quantification of secreted, intracellular, or cell membrane proteins. For
biomarker detection, AlphaLISA is mainly employed as a sandwich immunoassay. A
biotinylated anti-analyte antibody binds the streptavidin donor bead while a
second anti-analyte antibody is conjugated to AlphaLISA acceptor beads. In the
presence of the analyte, beads come into close proximity. Donor bead excitation
releases singlet oxygen molecules that transfer energy to the acceptor beads
with light emission at 615 nm (fig.). Alternatively, competition immunoassays
can also be adapted.

Cell-based assays
Here
described five cell-based readout technologies that have found general
application in many types of assay, namely fluorometric methods, reporter
gene assays, yeast complementation assays, high-throughput electrophysiology
assays and more recently label free detection platforms.
Fluorometric assays
Fluorometric assays
are widely used to monitor changes in the intracellular
concentration of ions or other constituents such as cAMP. A range of
fluorescent dyes has been developed which have the property of forming
reversible complexes with ions such as Ca2+ or Tl+ (as a surrogate for K+).
Their fluorescent emission intensity changes when the complex is formed, thereby
allowing changes in the free intracellular ion concentration to be monitored,
for example in response to activation or block of membrane receptors or ion
channels, Other membrane-bound dyes are available whose fluorescence signal
varies according to the cytoplasmic or mitochondrial membrane potential.
Membrane-impermeable dyes which bind to intracellular structures can be used to
monitor cell death, as only dying cells with leaky membranes are stained. In
addition to dyes, ion-sensitive proteins such as the jellyfish photo-protein
aequorin (see below), which emits a strong fluorescent signal when
complexed with Ca2+, can also be used to monitor changes in [Ca2+]i. Cell lines
can be engineered to express this protein, or it can be introduced by
electroporation. Such methods find many applications in cell biology,
particularly when coupled with confocal microscopy to achieve a high level of
spatial resolution. For HTS applications, the development of the Fluorescence
Imaging Plate Reader (FLIPR™, Molecular Devices Inc.,), allowing the
simultaneous application of reagents and test compounds to multiwell plates and
the capture of the fluorescence signal from each well was a key advance in
allowing cellular assays to be utilized in the HTS arena. Early instruments
employed an argon laser to deliver the excitation light source with the emission
measured using a CCD imaging device. In more recent models the laser has been
replaced with an LED light source (www.moleculardevices.com)
and overcomes some of the logistical considerations for deploying these
instruments in some laboratories. Repeated measurements can be made at intervals
of less than 1 s, to determine the kinetics of the cellular response, such as
changes in [Ca2+]i or membrane potential, which are often short-lasting, so that
monitoring the time profile rather than taking a single snapshot measurement is
essential.
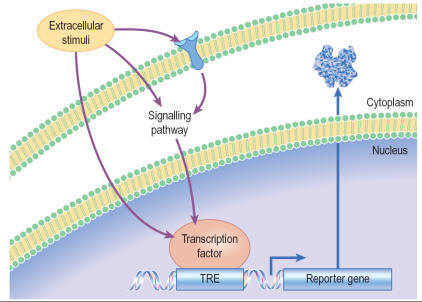
Reporter gene assays
Gene expression in transfected eukaryotic cells can be quantified
by linking a promoter sequence to a reporter gene, whose level of expression is
readily monitored, and reflects the degree of activation or inhibition of the
promoter. Compounds activating or inhibiting the promoter itself, or
interfering with a signal pathway connected to that promoter, can thus be
detected. By using two different reporter constructs e.g. firefly and Renilla
luciferase, different targets can be screened simultaneously. The principle
of a reporter gene assay for GPCR activity, based on luciferase, is shown in
Figure.
Reporter readouts can also be duplexed with more immediate readouts of cell
signalling, such as calcium sensitive dyes, to reduce the false positive
liability associated with using a single assay readout. Commonly used reporter
genes are CAT (chloramphenicol acetyltransferase), GAL (β-galactosidase), LAC
(β-lactamase), LUC (luciferase) and GFP (green fluorescence protein), usually
employing a colorimetric or fluorescent readout and each having relative merits.
The number of reporter genes is dwarfed compared to the range of promoters that
can be employed in this format, covering a diversity of signalling events.
Whilst having been widely deployed in the drug discovery process there are
several limitations of reporter gene technology, not least because of the
measurement of a response distal to the ligand interaction and the longer
compound incubation times required, increasing the potential for cytotoxic
events.
Yeast complementation assay
Yeast is a
well-characterized organism for investigating mammalian systems, and the yeast
two-hybrid assay is a powerful genetic screening technique for measuring the
protein–protein and protein–DNA interactions that underlie many cellular
control mechanisms. Widely applied in cell and systems biology to study the
binding of transcription factors at the sequence level, it can also be used to
screen small molecules for their interference with specific protein–protein and
protein–DNA interactions, and has recently been adapted for other types of
drug–target interactions. Conventional in vitro measurements, such as
immunoprecipitation or chromatographic co-precipitation, require the interacting
proteins in pure form and at high concentrations, and therefore are often of
limited use.
The yeast two-hybrid system uses two separated peptide domains of
transcription factors: a DNA-specific binding part (DNB) and a transcription
activation domain (AD). The DNB moiety is coupled to one protein (the ‘bait’),
and the AD moiety to another (the ‘prey’). If the prey protein binds to the bait
protein, the AD moiety is brought into close association with the reporter gene,
which is thereby activated, producing a product (e.g. GAL or LAC, as described
above, or an enzyme which allows the yeast to grow in the presence of
cycloheximide). The addition of a test compound that blocks the specific
protein–protein interaction prevents activation of the reporter gene. The bait
and prey proteins contained domains of two different channel subunits which need
to associate to form a functional channel.
High throughput electrophysiology assays
The
progression of ion channels, and in particular voltage-gated ion channels, as
druggable targets using screening approaches was, until recently, severely
limited by the throughput of conventional electrophysiology techniques and lack
of suitable higher throughput assay platforms. Although fluorescence methods
using membrane potential sensitive dyes such as DiBAC4(3) and the FLIPR™
variants of this and the FRET based voltage sensor probes were widely used, the
methodology could not provide accurate voltage control and the temporal
resolution of the evoked responses was poor. The introduction of planar
patch-clamp instruments, particularly systems such as IonWorks Quattro which
record using multihole planar substrate consumables has to a certain extent
overcome the throughput hurdle. The operating principle of this instrument is
shown in
Figure and whilst the data point generation is not as high throughput so as
to compete with fluorescence methods (a maximum of approximately 3000 data
points per day per instrument compared with > 20 000 per day for a FLIPR™) it is
sufficient for screening of targeted libraries, diverse compound decks up to
around 100 000 compounds and the confirmation of a large number of hits
identified in less physiologically relevant platforms.

Label free detection platforms
The current
drive for the drug discovery process is to move towards as physiologically
relevant systems as possible and away from target overexpression in heterologous
expression systems and, in the case of G-protein coupled receptors, to avoid
the use of promiscuous G-proteins where possible. The downside to this is that
endogenous receptor expression levels tend to be lower and therefore more
sensitive detection methods are required. Also, for the study of targets where
the signalling mechanism is unknown, e.g. orphan GPCRs, multiple assay systems
would need to be developed which would be time consuming and costly.
Consequently, the application of assay platforms which detect gross cellular
responses, usually cell morphology due to actin cytoskeleton remodelling, to
physiological stimuli have been developed. These fall into two broad categories,
those that detect changes in impedance through cellular dielectric spectroscopy,
e.g. CellKey™, Molecular Devices Corporation; xCelligence, Roche Diagnostics or
the use of optical biosensors, e.g. Epic™, Corning Inc; or Octect, Fortebio. The
application of these platforms in the HTS arena is still in its infancy largely
limited by throughput and relatively high cost per data point compared to
established methods. However, the assay development time is quite short, a
single assay may cover a broad spectrum of cell signalling events and these
methods are considered to be more sensitive than many existing methods enabling
the use of endogenous receptor expression and even the use of primary cells in
many instances.
High content screening
High content
screening (HCS) is a further development of cell-based screening in which
multiple fluorescence readouts are measured simultaneously in intact cells by
means of imaging techniques. Repetitive scanning provides temporally and
spatially resolved visualization of cellular events. HCS is suitable for
monitoring such events as nuclear translocation, apoptosis, GPCR activation,
receptor internalization, changes in [Ca2+]i, nitric oxide production,
apoptosis, gene expression, neurite outgrowth and cell viability. The aim is to
quantify and correlate drug effects on cellular events or targets by
simultaneously measuring multiple signals from the same cell population,
yielding data with a higher content of biological information than is provided
by single-target screens. Current instrumentation is based on automated digital
microscopy and flow cytometry in combination with hard and software systems for
the analysis of data. Within the configuration a fluorescence-based laser
scanning plate reader (96, 384- or 1536-well format), able to detect
fluorescent structures against a less fluorescent background, acquires
multicolour fluorescence image datasets of cells at a preselected spatial
resolution. The spatial resolution is largely defined by the instrument
specification and whether it is optical confocal or widefield. Confocal imaging
enables the generation of high-resolution images by sampling from a thin
cellular section and rejection of out of focus light; thus giving rise to
improved signal : noise compared to the more commonly applied epi-fluorescence
microscopy. There is a powerful advantage in confocal imaging for applications
where subcellular localization or membrane translocation needs to be measured.
However, for many biological assays, confocal imaging is not ideal e.g. where
there are phototoxicity issues or the applications have a need for a larger
focal depth. HCS relies heavily on powerful image pattern recognition software
in order to provide rapid, automated and unbiased assessment of experiments. The
concept of gathering all the necessary information about a compound at one go
has obvious attractions, but the very sophisticated instrumentation and software
produce problems of reliability. Furthermore, the principle of ‘measure
everything and sort it out afterwards’ has its drawbacks: interpretation of such
complex datasets often requires complex algorithms and significant data storage
capacity. Whilst the complexity of the analysis may seem daunting, high content
screening allows the study of complex signalling events and the use of
phenotypic readouts in highly disease relevant systems. However, such analysis
is not feasible for large number of compounds and unless the technology is the
only option for screening in most instances HCS is utilized for more detailed
study of lead compounds once they have been identified.
Biophysical methods in high-throughput screening
Conventional bioassay-based screening remains a mainstream
approach for lead discovery. However, during recent years alternative
biophysical methods such as nuclear magnetic resonance (NMR), surface plasmon
resonance (SPR) and X-ray crystallography have been developed and/or adapted for
drug discovery. Usually in assays whose main purpose is the detection of
low-affinity low-molecular-weight compounds in a different approach to
high-throughput screening, namely fragment-based screening. Hits from HTS
usually already have drug-like properties, e.g. a molecular weight of ~ 300 Da.
During the following lead optimization synthesis programme an increase in
molecular weight is very likely, leading to poorer drug-like properties with
respect to solubility, absorption or clearance. Therefore, it may be more
effective to screen small sets of molecular fragments (<10 000) of lower
molecular weight (100–250 Da) which can then be chemically linked to generate
high-affinity drug-like compounds. Typically, such fragments have much weaker
binding affinities than drug-like compounds and are outside the sensitivity
range of a conventional HTS assay. NMR-, SPR- or X-ray crystallography-based
assays are better suited for the identification of weak binders as these
methodologies lend themselves well to the area of fragment based screening. As
the compound libraries screened are generally of limited size throughput is less
important than sensitive detection of low-affinity interactions. Once the
biophysical interactions are determined, further X-ray protein crystallographic
studies can be undertaken to understand the binding mode of the fragments and
this information can then be used to rapidly drive the fragment-to-hit.
Pharmacology
Typically, when a molecular target has been selected, and lead
compounds have been identified which act on it selectively, and which are
judged to have ‘drug-like’ chemical attributes (including suitable
pharmacokinetic properties), the next stage is a detailed pharmacological
evaluation. This means investigation of the effects, usually of a small number
of compounds, on a range of test systems, up to and including whole animals, to
determine which, if any, is the most suitable for further development (i.e. for
nomination as a drug candidate). Pharmacological evaluation typically involves
the following:
•
Selectivity screening,
consisting of in vitro tests on a broad range of possible drug targets to
determine whether the compound is sufficiently selective for the chosen target
to merit further investigation
•
Pharmacological profiling, aimed at evaluating in isolated tissues or normal animals the
range of effects of the test compound that might be relevant in the clinical
situation. Some authorities distinguish between primary pharmacodynamic
studies, concerning effects related to the selected therapeutic target (i.e.
therapeutically relevant effects), and secondary pharmacodynamic studies,
on effects not related to the target (i.e. side effects). At the laboratory
level the two are often not clearly distinguishable, and the borderline between
secondary pharmacodynamic and safety pharmacology studies (see below) is also
uncertain. Nevertheless, for the purposes of formal documentation, the
distinction may be useful
•
Testing in animal models of disease
to determine whether the compound is likely to produce
therapeutic benefit
•
Safety
pharmacology,
consisting of a series of standardized animal tests aimed at revealing
undesirable side effects, which may be unrelated to the primary action of the
drug.
The
pharmacological evaluation of lead compounds does not in general follow a
clearly defined path, and often it has no clearcut endpoint but will vary
greatly in its extent, depending on the nature of the compound, the questions
that need to be addressed and the inclinations of the project team. Directing
this phase of the drug discovery project efficiently, and keeping it focused on
the overall objective of putting a compound into development, is one of the
trickier management tasks. It often happens that unexpected, scientifically
interesting data are obtained which beg for further investigation even though
they may be peripheral to the main aims of the project. From the scientists’
perspective, the prospect of opening up a new avenue of research is highly
alluring, whether the work contributes directly to the drug discovery aims or
not. In this context, project managers need to bear in mind the question: Who
needs the data and why? – a question which may seem irritatingly silly to a
scientist in academia but totally obvious to the commercial mind. The same
principles apply, of course, to all parts of a drug discovery and development
project, but it tends to be at the stage of pharmacological evaluation that
conflicts first arise between scientific aspiration and commercial need.
An
important principle in pharmacological evaluation is the use of a hierarchy
of test methods, covering the range from the most reductionist tests on
isolated molecular targets to much more elaborate tests of integrated
physiological function. Establishing and validating such a series of tests
appropriate to the particular target and indication being addressed is one of
the most important functions of pharmacologists in the drug discovery team. In
general, assays become more complicated, slow and expensive, and more demanding
of specialist skills as one moves up this hierarchy.
The
strengths and weaknesses of these test systems are summarized in
Table.
Pharmacological characterization of a candidate compound often has to take into
account active metabolites, based on information from drug metabolism and
pharmacokinetics (DMPK) studies. If a major active metabolite is identified, it
will be necessary to synthesize and test it in the same way as the parent
compound in order to determine which effects (both wanted and unwanted) relate
to each. Particular problems may arise if the metabolic fate of the compound
shows marked species differences, making it difficult to predict from animal
studies what will happen in humans.

Although most of the work involved in pharmacological characterization of a
candidate drug takes place before clinical studies begin, it does not normally
end there. Both ongoing toxicological studies and early trials in man may reveal
unpredicted effects that need to be investigated pharmacologically, and so the
discovery team needs to remain actively involved and be able to perform
experiments well into the phase of clinical development. They cannot simply
wave the compound goodbye once the discovery phase is completed.
SCREENING FOR SELECTIVITY
The selectivity of a compound for the chosen molecular target
needs to be assessed at an early stage. Compounds selected for their potency,
for example on a given amine receptor, protease, kinase, transporter or ion
channel, are very likely to bind also to related – or even unrelated – molecular
targets, and thereby cause unwanted side effects. Selectivity is, therefore, as
important as potency in choosing potential development candidates, and a
‘selectivity screen’ is usually included early in the project. The range of
targets included in such a screen depends very much on the type of compound and
the intended clinical indication. Ligands for monoamine receptors and
transporters form a large and important group of drugs, and several contract
research organizations (e.g. CEREP, MDL) offer a battery of assays – mainly
binding assays, but also a range of functional assays – designed to detect
affinity for a wide range of receptors, transporters and channels. In the field
of monoamine receptors, for example, it is usually important to avoid compounds
that block or activate peripheral muscarinic receptors, adrenergic receptors or
histamine (particularly H1) receptors,
because of the side effects that are associated with these actions, and a
standard selectivity test battery allows such problems to be discovered early.
Recently, several psychotropic and anti-infective drugs have been withdrawn
because of sudden cardiac deaths, probably associated with their ability to
block a particular type of potassium channel (known as the hERG channel)
in myocardial cells. This activity can be detected by electrophysiological
measurements on isolated myocardial cells, and such a test is now usually
performed at an early stage of development of drugs of the classes implicated in
this type of adverse reaction.
Interpretation of binding assays
Binding assays, generally with membrane preparations made from
intact tissues or receptor-expressing cell lines, are widely used in drug
discovery projects because of their simplicity and ease of automation. Detailed
technical manuals describing the methods used for performing and analysing drug
binding experiments are available (Keen, 1999;
Vogel, 2002). Generally, the aim of the assay is to determine the
dissociation constant, KD,
of the test compound, as a measure of its affinity for the receptor. In most
cases, the assay (often called a displacement assay) measures the ability
of the test compound to inhibit the binding of a high-affinity radioligand which
combines selectively with the receptor in question, correction being made for
‘non-specific’ binding of the radioligand. In the simplest theoretical case,
where the radioligand and the test compound bind reversibly and competitively to
a homogeneous population of binding sites, the effect of the test ligand on the
amount of the radioligand specifically bound is described by the simple
mass-action equation:
![]() (1)
(1)
where B = the amount of radioligand bound, after
correcting for non-specific binding, Bmax
= the maximal amount of radioligand bound, i.e. when sites are
saturated, [A] = radioligand concentration, KA
= dissociation constant for the radioligand, [L] = test ligand
concentration, and KL =
dissociation constant for the test ligand.
By
testing several concentrations of L at a single concentration of A, the
concentration, [L]50, needed for 50%
inhibition of binding can be estimated. By rearranging equation 1, KL is given by:
![]() (2)
(2)
This is often known as the Cheng–Prusoff equation, and is widely
used to calculate KL
when [L]50, [A] and
KA are known. It
is important to realize that the Cheng–Prusoff equation applies only (a) at
equilibrium, (b) when the interaction between A and L is strictly competitive,
and (c) when neither ligand binds cooperatively. However, an [L]50 value can be
measured for any test compound that inhibits the binding of the radioligand by
whatever mechanism, irrespective of whether equilibrium has been reached.
Applying the Cheng–Prusoff equation if these conditions are not met can yield
estimates of KL that are
quite meaningless, and so it should strictly be used only if the conditions
have been shown experimentally to be satisfied – a fairly laborious process.
Nevertheless, Cheng–Prusoff estimates of ligand affinity constants are often
quoted without such checks having been performed. In most cases it would be more
satisfactory to use the experimentally determined [L]50
value as an operational measure of potency. A further important
caveat that applies to binding studies is that they are often performed under
conditions of low ionic strength, in which the sodium and calcium concentrations
are much lower than the physiological range. This is done for technical reasons,
as low [Na+] commonly increases both the
affinity and the Bmax of
the radioligand, and omitting [Ca2+]
avoids clumping of the membrane fragments. Partly for this reason, ligand
affinities estimated from binding studies are often considerably higher than
estimates obtained from functional assays (Hall, 1992), although the effect is not consistent, presumably because
ionic bonding, which will be favoured by the low ionic strength medium,
contributes unequally to the binding of different ligands. Consequently, the
correlation between data from binding assays and functional assays is often
rather poor (see below).
Figure
shows
data obtained independently on 5HT3 and 5HT4 receptors; in both cases the
estimated KD values for binding are on average about 10 times lower than
estimates from functional assays, and the correlation is very poor.
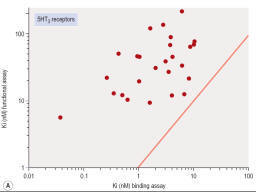
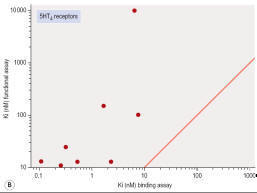
Figure
- Correlation of binding and functional data for 5HT receptor ligands. (A) 5HT3
receptors, (B) 5HT4
receptors.
Pharmacological profiling
Pharmacological profiling aims to determine the pharmacodynamic
effects of the new compound – or more often of a small family of compounds – on
in vitro model systems, e.g. cell lines or isolated tissues, normal animals, and
animal models of disease. The last of these is particularly important, as it is
intended to give the first real pointer to therapeutic efficacy as distinct from
pharmacodynamic activity. It is valuable to assess the activity of the
compounds in a series of assays representing increasingly complex levels of
organization. The choice of test systems depends, of course, on the nature of
the target. For example, characterization of a novel antagonist of a typical
G-protein-coupled receptor might involve the following:
•
Ligand-binding assay on membrane fragments from a cell line expressing the
cloned receptor
•
Inhibition of agonist activity in a cell line, based on a functional readout
(e.g. raised intracellular calcium)
•
Antagonism of a selective agonist in an isolated tissue (e.g. smooth muscle,
cardiac muscle). Such assays will normally be performed with non-human tissue,
and so interspecies differences in the receptor need to be taken into account.
Sometimes specific questions have to be asked about effects on human tissues for
particular compounds and then collecting viable tissues to use becomes a major
challenge
•
Antagonism of the response (e.g. bronchoconstriction, vasoconstriction,
increased heart rate) to a selective receptor agonist in vivo. Prior knowledge
about species specificity of the agonist and antagonist is important at this
stage.
Pharmacological profiling is designed as a hypothesis-driven programme of work,
based on the knowledge previously gained about the activity of the compound on
its specific target or targets. In this respect it differs from safety
pharmacology (see below), which is an open-minded exercise designed to detect
unforeseen effects. The aim of pharmacological profiling is to answer the
following questions:
•
Do the
molecular and cellular effects measured in screening assays actually give rise
to the predicted pharmacological effects in intact tissues and whole animals?
•
Does
the compound produce effects in intact tissues or whole animals not associated
with actions on its principal molecular target?
•
Is
there correspondence between the potency of the compound at the molecular level,
the tissue level and the whole animal level?
•
Do the
in vivo potency and duration of action match up with the pharmacokinetic
properties of the compound? What happens if the drug is given continuously or
repeatedly to an animal over the course of days or weeks?
•
Does
it lose its effectiveness, or reveal effects not seen with acute administration?
Is there any kind of ‘rebound’ after effect when it is stopped?
In vitro profiling
Measurements on
isolated tissues
Studies on
isolated tissues have been a mainstay of pharmacological methodology ever since
the introduction of the isolated organ bath by Magnus early in the 20th century.
The technique is extremely versatile and applicable to studies on smooth muscle
(e.g. gastrointestinal tract, airways, blood vessels, urinary tract, uterus,
biliary tract, etc.) as well as cardiac and striated muscle, secretory
epithelia, endocrine glands, brain slices, liver slices, and many other
functional systems. In most cases the tissue is removed from a freshly killed or
anaesthetized animal and suspended in a chamber containing warmed oxygenated
physiological salt solution. With smooth muscle preparations the readout is
usually mechanical (i.e. tension, recorded with a simple strain gauge). For
other types of preparation, various electrophysiological or biochemical
readouts are often used.
Studies of this kind have the advantage that they are performed
on intact normal tissues, as distinct from isolated enzymes or other proteins.
The recognition molecules, signal transduction machinery and the mechanical or
biochemical readout are assumed to be a reasonable approximation to the normal
functioning of the tissue. There is abundant evidence to show that tissue
responses to GPCR activation, for example, depend on many factors, including the
level of expression of the receptor, the type and abundance of the G proteins
present in the cell, the presence of associated proteins such as receptor
activity-modifying proteins (RAMPs), the state of phosphorylation of various
constituent proteins in the signal transduction cascade, and so on. For
compounds acting on intracellular targets, functional activity depends on
permeation through the membrane, as well as affinity for the target. For these
reasons – and probably also for others that are not understood – the results of
assays on isolated tissues often differ significantly from results found with
primary screening assays. The discrepancy may simply be a quantitative one,
such that the potency of the ligand does not agree in the two systems, or it may
be more basic. For example, the pharmacological efficacy of a receptor
ligand, i.e. the property that determines whether it is a full agonist, a
partial agonist, or an antagonist, often depends on the type of assay used, and
this may have an important bearing on the selection of possible development
compounds. Examples that illustrate the poor correlation that may exist between
measurements of target affinity in cell-free assay systems, and functional
activity in intact cell systems, are shown in
Figures.
Figure shows the relationship between binding and functional assay data for
5HT3 and 5HT4 receptor antagonists. In both cases, binding assays overestimate
the potency in functional assays by a factor of about 10 (see above), but more
importantly, the correlation is poor, despite the fact that the receptors are
extracellular, and so membrane penetration is not a factor.
Figure
shows data on tyrosine kinase inhibitors, in which activity
against the isolated enzyme is plotted against inhibition of tyrosine
phosphorylation in intact cells, and inhibition of cell proliferation for a
large series of compounds. Differences in membrane penetration can account for
part of the discrepancy between enzyme and cell-based data, but the correlation
between intracellular kinase inhibition and blocking of cell proliferation is
also weak, which must reflect other factors.
It is worth noting that these examples come from very successful
drug discovery projects. The quantitative discrepancies that we have
emphasized, though worrying to pharmacologists, should not therefore be a
serious distraction in the context of a drug discovery project. A very wide
range of physiological responses can be addressed by studies on isolated
tissues, including measurements of membrane excitability, synaptic function,
muscle contraction, cell motility, secretion and release of mediators,
transmembrane ion fluxes, vascular resistance and permeability, and epithelial
transport and permeability. This versatility and the relative technical
simplicity of many such methods are useful attributes for drug discovery.
Additional advantages are that concentration–effect relationships can be
accurately measured, and the design of the experiments is highly flexible,
allowing rates of onset and recovery of drug effects to be determined, as well
as measurements of synergy and antagonism by other compounds, desensitization
effects, etc. The main shortcomings
of isolated tissue pharmacology are (a) that tissues normally have to be
obtained from small laboratory animals, rather than humans or other primates;
and (b) that preparations rarely survive for more than a day, so that only
short-term experiments are feasible.

Figure
Correlation of cellular activity of EGFR receptor kinase inhibitors with enzyme
inhibition.
In vivo profiling
As already mentioned, experiments on animals have several
drawbacks. They are generally time-consuming, technically demanding and
expensive. They are subject to considerable ethical and legal constraints, and
in some countries face vigorous public opposition. For all these reasons, the
number of experiments is kept to a bare minimum, and experimental variability is
consequently often a problem. Animal experiments must, therefore, be used very
selectively and must be carefully planned and designed so as to produce the
information needed as efficiently as possible. In the past, before
target-directed approaches were the norm, routine in vivo testing was often used
as a screen at a very early stage in the drug discovery process, and many
important drugs (e.g. thiazide diuretics, benzodiazepines, ciclosporin) were
discovered on the basis of their effects in vivo. Nowadays, the use of in vivo
methods is much more limited, and will probably decline further in response to
the pressures on time and costs, as alternative in vitro and in silico methods
are developed, and as public attitudes to animal experimentation harden. An
additional difficulty is the decreasing number of pharmacologists trained to
perform in vivo studies1.
Imaging
technologies are increasingly being used for pharmacological studies on whole
animals. Useful techniques include magnetic resonance imaging (MRI), ultrasound
imaging, X-ray densitometry tomography, positron emission tomography (PET) and
others. They are proving highly versatile for both structural measurements (e.g.
cardiac hypertrophy, tumour growth) and functional measurements (e.g. blood
flow, tissue oxygenation). Used in conjunction with radio-active probes, PET can
be used for studies on receptors and other targets in vivo. Many of these
techniques can also be applied to humans, providing an important bridge between
animal and human pharmacology. Apart from the special facilities and equipment
needed, currently the main drawback of imaging techniques is the time taken to
capture the data, during which the animal must stay still, usually necessitating
anaesthesia. With MRI and PET, which are currently the most versatile imaging
techniques, data capture normally takes a few minutes, so they cannot be used
for quick ‘snapshots’ of rapidly changing events.
A particularly important role for in vivo experiments is to
evaluate the effects of long-term drug administration on the intact organism.
‘Adaptive’ and ‘rebound’ effects (e.g. tolerance, dependence, rebound
hypertension, delayed endocrine effects, etc.) are often produced when drugs are
given continuously for days or weeks. Generally, such effects, which involve
complex physiological interactions, are evident in the intact functioning
organism but are not predictable from in vitro experiments.

The
programme of in vivo profiling studies for characterization of a candidate drug
depends very much on the drug target and therapeutic indication. A comprehensive
catalogue of established in vivo assay methods appropriate to different types of
pharmacological effect is given by
Vogel (2002). Charting the appropriate course through the plethora of
possible studies that might be performed to characterize a particular drug can
be difficult.
A typical example of pharmacological profiling is summarized in
Box. The studies were carried out as part of the recent development
of a cardiovascular drug, beraprost. Beraprost is a stable analogue of
prostaglandin I2 (PGI2) which acts on PGI2 receptors of platelets and blood
vessels, thereby inhibiting platelet aggregation (and hence thrombosis) and
dilating blood vessels. It is directed at two therapeutic targets, namely
occlusive peripheral vascular disease and pulmonary hypertension (a serious
complication of various types of cardiovascular disease, drug treatment or
infectious diseases), resulting in hypertrophy and often contractile failure of
the right ventricle. The animal studies were, therefore, directed at measuring
changes (reduction in blood flow, histological changes in vessel wall)
associated with peripheral vascular disease, and with pulmonary hypertension. As
these are progressive chronic conditions, it was important to establish that
long-term systemic administration of beraprost was effective in retarding the
development of the experimental lesions, as well as monitoring the acute
pharmacodynamic effects of the drug.
Species differences
It is
important to take species differences into account at all stages of
pharmacological profiling. For projects based on a defined molecular target –
nowadays the majority – the initial screening assay will normally involve the
human isoform. The same target in different species will generally differ in its
pharmacological specificity; commonly, there will be fairly small quantitative
differences, which can be allowed for in interpreting pharmacological data in
experimental animals, but occasionally the differences are large, so that a
given class of compounds is active in one species but not in another. An example
is shown in
Figure, which compares the activities of a series of bradykinin
receptor antagonists on cloned human and rat receptors. The complete lack of
correlation means that, for these compounds, tests of functional activity in the
rat cannot be used to predict activity in man.
Species differences are, in fact, a major complicating factor at
all stages of drug discovery and preclinical development. The physiology of
disease processes such as inflammation, septic shock, obesity, atherosclerosis,
etc., differs markedly in different species. Most importantly (see Chapter 10),
drug metabolism often differs, affecting the duration of action, as well as the
pattern of metabolites, which can in turn affect the observed pharmacology and
toxicity.
Species differences are, of course, one of the main arguments used by animal
rights activists in opposing the use of animals for the purpose of drug
discovery. Their claim – misleading when examined critically (see Understanding
Animal Research website) – is that animal data actually represent disinformation
in this context. While being aware of the pitfalls, we should not lose sight of
the fact that non-human data, including in vivo experiments, have actually been
an essential part of every major drug discovery project to date. The growing use
of transgenic animal models has led to an increase, rather than a decrease, in
animal experimentation, as even breeding such animals is counted as an
experiment for statistical purposes.
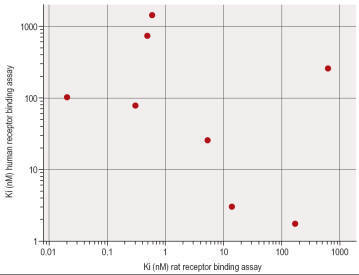
Figure - Species differences in
bradykinin B2
receptors.
ANIMAL MODELS OF DISEASE
The animal models discussed earlier were used to investigate the
pharmacodynamic effects of the drug and to answer the question: How do the
effects observed at the molecular and cellular levels of organization translate
into physiological effects in the whole animal?
The next, crucial, question is: Can these physiological effects
result in therapeutic benefit? Animal experiments can never answer this
conclusively – only clinical trials can do that – but the use of animal models
of human disease provides a valuable link in the chain of evidence, and there is
strong pressure on drug discovery teams to produce data of this sort as a basis
for the important decision to test a new compound in man. Despite the immense
range and diversity of animal models that have been described, this is often the
most problematic aspect of a drug discovery project, particularly where a novel
target or mechanism is involved, so that there is no mechanistic precedent among
established drugs. The magnitude of the difficulties varies considerably among
different therapeutic areas. Many inflammatory conditions, for example, are
straightforward to model in animals, as are some cancers. Animal models of
hypertension generally predict very well the ability of compounds to lower blood
pressure in man. Endocrine disorders involving over- or under secretion of
particular hormones can also be simply modelled in animals. Psychiatric
disorders are much more difficult, as the symptoms that characterize them are
not observable in animals. In most therapeutic areas there are certain
disorders, such as migraine, temporal lobe epilepsy, asthma or irritable bowel
syndrome, for which animal models, if they exist at all, are far from
satisfactory in predicting clinical efficacy.
Here we consider, with a few selected examples, the main
experimental approaches to generating animal models, and the criteria against
which their ‘validity’ as models of human disease need to be assessed.
Types of animal
model
Animal
models of disease can be divided broadly into acute and chronic physiological
and pharmacological models, and genetic models.
Acute physiological and pharmacological models
are intended to mimic certain aspects of the clinical disorder. There are many
examples, including:
•
Seizures induced by electrical stimulation of the brain as a
model for epilepsy (see below)
•
Histamine-induced bronchoconstriction as a model for asthma
•
The hotplate test for analgesic drugs as a model for pain
•
Injection of lipopolysaccharide (LPS) and cytokines as a model
for septic shock
•
The elevated maze test as a model for testing anxiolytic drugs.
Chronic physiological or pharmacological models
involve the use of drugs or physical interventions to induce an ongoing
abnormality similar to the clinical condition. Examples include:
•
The use of alloxan to inhibit insulin secretion as a model for
Type I diabetes
•
Procedures for inducing brain or coronary ischaemia as models for
stroke and ischaemic heart disease
‘Kindling’ and other procedures for inducing ongoing seizures as
models for epilepsy
•
Self-administration of opiates, nicotine or other drugs as a
model for drug-dependence
•
Cholesterol-fed rabbits as a model for hypercholesterolaemia and
atherosclerosis
•
Immunization with myelin basic protein as a model for multiple
sclerosis
•
Administration of the neurotoxin MPTP, causing degeneration of
basal ganglia neurons as a model of Parkinson’s disease
•
Transplantation of malignant cells into immunodeficient animals
to produce progressive tumours as a model for certain types of cancer.
Details of these and many other examples of physiological and
pharmacological models can be found in
Vogel (2002).
As discussed above, species differences need to be taken into account in the
selection of animal models, and in the interpretation of results. In septic
shock, for example, rodents show a much larger elevation of nitric oxide
(NO) metabolites than do humans, and respond well to NO synthesis inhibitors,
which humans do not. Rodents and rabbits transgenically engineered to favour
cholesterol deposition nevertheless develop atherosclerosis only when fed
high-cholesterol diets, whereas humans often do so even on low-cholesterol
diets. Genetically obese mice are deficient in the hormone leptin and
lose weight when treated with it, whereas obese humans frequently have high
circulating leptin concentrations and do not respond to treatment with it. It is
often not clear whether such discrepancies reflect inherent species differences,
or simply failure of the model to replicate satisfactorily the predominant
human disease state.
Genetic models
There are
many examples of spontaneously occurring animal strains that show abnormalities
phenotypically resembling human disease. In addition, much effort is going into
producing transgenic strains with deletion or over-expression of specific genes,
which also exhibit disease-like phenotypes.
Long before genetic mapping became possible, it was realized that
certain inbred strains of laboratory animal were prone to particular disorders,
examples being spontaneously hypertensive rats, seizure-prone dogs, rats
insensitive to antidiuretic hormone (a model for diabetes insipidus), obese mice
and mouse strains exhibiting a range of specific neurological deficits. Many
such strains have been characterized (Jackson Laboratory website,
www.jaxmice.jax.org) and are
commercially available, and are widely used as models for testing drugs.
The
development of transgenic technology has allowed inbred strains to be produced
that over- or under-express particular genes. In the simplest types, the gene
abnormality is present throughout the animal’s life, from early development
onwards, and throughout the body. More recent technical developments allow much
more control over the timing and location of the transgene effect. For reviews
of transgenic technology and its uses in drug discovery.
The
genetic analysis of disease-prone animal strains, or of human families affected
by certain diseases, has in many cases revealed the particular mutation or
mutations responsible, thus pointing the way to new transgenic models. Several
diseases associated with single-gene mutations, such as cystic fibrosis
and Duchenne muscular dystrophy, have been replicated in transgenic
mouse strains. Analysis of the obese mouse strain led to the identification of
the leptin gene, which is mutated in the ob/ob mouse strain, causing the
production of an inactive form of the hormone and overeating by the mouse.
Transgenic animals closely resembling ob/ob mice have been produced by
targeted inactivation of the gene for leptin or its receptor. Another example is
the discovery that a rare familial type of Alzheimer’s disease is associated
with mutations of the amyloid precursor protein (APP). Transgenic mice
expressing this mutation show amyloid plaque formation characteristic of the
human disease. This and other transgenic models of Alzheimer’s disease
represent an important tool for drug discovery, as there had hitherto been no
animal model reflecting the pathogenesis of this disorder.
The
number of transgenic animal models, mainly mouse, that have been produced is
already large and is growing rapidly. Creating and validating a new disease
model is, however, a slow business. Although the methodology for generating
transgenic mice is now reliable and relatively straightforward, it is both
time-consuming and labour-intensive. The first generation of transgenic animals
are normally hybrids, as different strains are used for the donor and the
recipient, and it is necessary to breed several generations by repeated
back-crossings to create animals with a uniform genetic background. This takes
1–2 years, and is essential for consistent results. Analysis of the phenotypic
changes resulting from the transgene can also be difficult and time-consuming,
as the effects may be numerous and subtle, as well as being slow to develop as
the animal matures. Despite these difficulties, there is no doubt that
transgenic disease models are playing an increasing part in drug testing, and
many biotechnology companies have moved into the business of developing and
providing them for this purpose. The fields in which transgenic models have so
far had the most impact are cancer, atherosclerosis and neurodegenerative
diseases, but their importance as drug discovery tools extends to all areas.
Producing transgenic rat strains proved impossible until recently, as embryonic
stem (ES) cells cannot be obtained from rats. Success in producing gene knockout
strains by an alternative method has now been achieved, and the use of
transgenic rats is increasing, this being the favoured species for
pharmacological and physiological studies in many laboratories.
The choice of model
Naturally
occurring diseases produce a variety of structural biochemical abnormalities,
and these are often displayed separately in animal models. For example, human
allergic asthma involves: (a) an immune response; (b) increased airways
resistance; (c) bronchial hyperreactivity; (d) lung inflammation; and (e)
structural remodelling of the airways. Animal models, mainly based on guinea
pigs, whose airways behave similarly to those of humans, can replicate each of
these features, but no single model reproduces the whole spectrum. The choice of
animal model for drug discovery purposes, therefore, depends on the therapeutic
effect that is being sought. In the case of asthma, existing bronchodilator
drugs effectively target the increased airways resistance, and steroids reduce
the inflammation, and so it is the other components for which new drugs are
particularly being sought. A similar need for a range of animal models covering
a range of therapeutic targets applies in many disease areas.
Validity criteria
Obviously an
animal model produced in a laboratory can never replicate exactly a spontaneous
human disease state, so on what basis can we assess its ‘validity’ in the
context of drug discovery?
Three types of validity criteria were originally proposed by
Willner (1984)
in connection with animal models of depression. These are:
•
Face validity
•
Construct validity
•
Predictive validity.
Face validity
refers to the
accuracy with which the model reproduces the phenomena (symptoms, clinical signs
and pathological changes) characterizing the human disease.
Construct validity
refers to the
theoretical rationale on which the model is based, i.e. the extent to which the
aetiology of the human disease is reflected in the model. A transgenic animal
model in which a human disease-producing mutation is replicated will have, in
general, good construct validity, even if the manifestations of the human
disorder are not well reproduced (i.e. it has poor face validity).
Predictive validity
refers to the
extent to which the effect of manipulations (e.g. drug treatment) in the model
is predictive of effects in the human disorder. It is the most pragmatic of the
three and the most directly relevant to the issue of predicting therapeutic
efficacy, but also the most limited in its applicability, for two main reasons.
First, data on therapeutic efficacy are often sparse or non-existent, because no
truly effective drugs are known (e.g. for Alzheimer’s disease, septic shock).
Second, the model may focus on a specific pharmacological mechanism, thus
successfully predicting the efficacy of drugs that work by that mechanism but
failing with drugs that might prove effective through other mechanisms. The
knowledge that the first generation of antipsychotic drugs act as dopamine
receptor antagonists enabled new drugs to be identified by animal tests
reflecting dopamine antagonism, but these tests cannot be relied upon to
recognize possible ‘breakthrough’ compounds that might be effective by other
mechanisms. Thus, predictive validity, relying as it does on existing
therapeutic knowledge, may not be a good basis for judging animal models where
the drug discovery team’s aim is to produce a mechanistically novel drug. The
basis on which predictive validity is judged carries an inevitable bias, as the
drugs that proceed to clinical trials will normally have proved effective in
the model, whereas drugs that are ineffective in the model are unlikely to have
been developed. As a result, there are many examples of tests giving ‘false
positive’ expectations, but very few false negatives, giving rise to a commonly
held view that conclusions from pharmacological tests tend to be overoptimistic.
Some examples
We conclude
this discussion of the very broad field of animal models of disease by
considering three disease areas, namely epilepsy, psychiatric disorders and
stroke. Epilepsy-like seizures can be produced in laboratory animals in many
different ways. Many models have been described and used successfully to
discover new anti-epileptic drugs (AEDs). Although the models may lack construct
validity and are weak on face validity, their predictive validity has proved to
be very good. With models of psychiatric disorders, face validity and construct
validity are very uncertain, as human symptoms are not generally observable in
animals and because we are largely ignorant of the cause and pathophysiology of
these disorders; nevertheless, the predictive validity of available models of
depression, anxiety and schizophrenia has proved to be good, and such models
have proved their worth in drug discovery. In contrast, the many available
models of stroke are generally convincing in terms of construct and face
validity, but have proved very unreliable as predictors of clinical efficacy.
Researchers in this field are ruefully aware that despite many impressive
effects in laboratory animals, clinical successes have been negligible.
Epilepsy models
The
development of antiepileptic drugs, from the pioneering work of Merritt and
Putnam, who in 1937 developed phenytoin, to the present day, has been highly
dependent on animal models involving experimentally induced seizures, with
relatively little reliance on knowledge of the underlying physiological,
cellular or molecular basis of the human disorder. Although existing drugs have
significant limitations, they have brought major benefits to sufferers from this
common and disabling condition – testimony to the usefulness of animal models in
drug discovery.
Human epilepsy is a chronic condition with many underlying
causes, including head injury, infections, tumours and genetic factors.
Epileptic seizures in humans take many forms, depending mainly on where the
neural discharge begins and how it spreads.
Some of the widely used animal models used in drug discovery are
summarized in
Table. The earliest models, namely the maximal electroshock (MES)
test and the pentylenetetrazol-induced seizure (PTZ) test, which are
based on acutely induced seizures in normal animals, are still commonly used.
They model the seizure, but without distinguishing its localization and spread,
and do not address either the chronicity of human epilepsy or its aetiology
(i.e. they score low on face validity and construct validity). But, importantly,
their predictive validity for conventional antiepileptic drugs in man is very
good, and the drugs developed on this basis, taken regularly to reduce the
frequency of seizures or eliminate them altogether, are of proven therapeutic
value. Following on from these acute seizure models, attempts have been made to
replicate the processes by which human epilepsy develops and continues as a
chronic condition with spontaneous seizures, i.e. to model epileptogenesis
by the use of models that show greater construct and face validity. This has
been accomplished in a variety of ways (Table)
in the hope that such models would be helpful in developing drugs capable of
preventing epilepsy. Such models have thrown considerable light on the
pathogenesis of epilepsy, but have not so far contributed significantly to the
development of improved antiepileptic drugs. Because there are currently no
drugs known to prevent epilepsy from progressing, the predictive validity of
epileptogenesis models remains uncertain.
Psychiatric disorders
Animal
models of psychiatric disorders are in general problematic, because in many
cases the disorders are defined by symptoms and behavioural changes unique to
humans, rather than by measurable physiological, biochemical or structural
abnormalities. This is true in conditions such as schizophrenia, Tourette’s
syndrome and autism, making face validity difficult to achieve. Depressive
symptoms, in contrast, can be reproduced to
some extent
in animal models, and face validity is therefore stronger. The aetiology of most
psychiatric conditions is largely unknown2,
making construct validity questionable.
Models are therefore chosen largely on the basis of predictive
validity, and suffer from the shortcomings mentioned above. Nonetheless, models
for some disorders, particularly depression, have proved very valuable in the
discovery of new drugs. Other disorders, such as autism and Tourette’s syndrome,
have proved impossible to model so far, whereas models for others, such as
schizophrenia, have been described but are of doubtful validity. The best
prediction of antipsychotic drug efficacy comes from pharmacodynamic models
reflecting blockade of dopamine and other monoamine receptors, rather than from
putative disease models, with the result that drug discovery has so far failed
to break out of this mechanistic straitjacket.
Stroke
Many experimental procedures have been devised to produce acute
cerebral ischaemia in laboratory animals, resulting in long-lasting neurological
deficits that resemble the sequelae of strokes in humans. Interest in this area
has been intense, reflecting the fact that strokes are among the commonest
causes of death and disability in developed countries, and that there are
currently no drugs that significantly improve the recovery process. Studies with
animal models have greatly advanced our understanding of the pathophysiological
events. Stroke is no longer seen as simple anoxic death of neurons, but rather
as a complex series of events involving neuronal depolarization, activation of
ion channels, release of excitatory transmitters, disturbed calcium homeostasis
leading to calcium overload, release of inflammatory mediators and nitric oxide,
generation of reactive oxygen species, disturbance of the blood–brain barrier
and cerebral oedema. Glial cells, as well as neurons, play an important role in
the process. Irreversible loss of neurons takes place gradually as this cascade
builds up, leading to the hope that intervention after the primary event –
usually thrombosis – could be beneficial. Moreover, the biochemical and cellular
events involve well-understood signalling mechanisms, offering many potential
drug targets, such as calcium channels, glutamate receptors, scavenging of
reactive oxygen species and many others. Ten years ago, on the basis of various
animal models with apparently good construct and face validity and a range of
accessible drug targets, the stage seemed to be set for major therapeutic
advances. Drugs of many types, including glutamate antagonists, calcium and
sodium channel blocking drugs, anti-inflammatory drugs, free radical scavengers
and others, produced convincing degrees of neuroprotection in animal models,
even when given up to several hours after the ischaemic event. Many clinical
trials were undertaken, with uniformly negative results. The only drug currently
known to have a beneficial – albeit small – effect is the biopharmaceutical
‘clot-buster’ tissue plasminogen activator (TPA), widely used to treat heart
attacks. Stroke models thus represent approaches that have revealed much about
pathophysiology and have stimulated intense efforts in drug discovery, but
whose predictive validity has proved to be extremely poor, as the drug
sensitivity of the animal models seems to be much greater than that of the human
condition. Surprisingly, it appears that whole-brain ischaemia models show
better predictive validity (i.e. poor drug responsiveness) than focal ischaemia
models, even though the latter are more similar to human strokes.
Zebrafish
The Zebrafish
is a vertebrate animal model with high genetic homology to humans that gathers
all these benefits simultaneously. They are often seen as a bridge
between in vitro and in vivo
models. Specifically, Zebrafish larvae
offer a viable and cost-effective model that supports High Content
Screening Assays without the ethical concerns typically associated with
in vivo studies, as they are not classified as such when used under 5-6
days post-fertilization (dpf). Given these attributes, Zebrafish are
increasingly becoming valuable in the Drug Discovery and Development process.
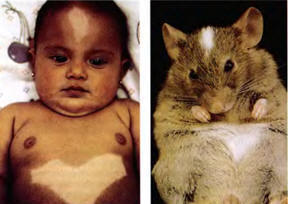
Mouse
Among mammals, the mouse is the most
suitable for genetic analysis, which is facilitated by the availability of its
complete genome sequence. Although the technical difficulties in studying mouse
genetics (compared, for example, to the genetics of yeasts or Drosophila) are
formidable, many mutations affecting mouse development have been identified.
Most important, recent advances in molecular biology have enabled the production
of genetically engineered mice in which specific mutant genes have been
introduced into the mouse germ line, allowing the functions of these genes to be
studied in the context of the whole animal. The suitability of the mouse as a
model for human development is indicated not only by the similarity of the mouse
and human genomes but also by the fact that mutations in homologous genes result
in similar developmental defects in both species; piebaldism is a striking
example.
Ex Vivo studies
Ex vivo,
meaning out of the living in Latin, refers to experiments or procedures
performed on tissues or organs extracted from a living organism and conducted
outside the original biological context. These experiments are conducted in a
laboratory setting, where the biological material is maintained in conditions
that mimic those found within the organism as closely as possible.
The architecture, including
cell-extracellular matrix (ECM) and cell-cell connections, is maintained,
granting that metabolic processes are closer to the in vivo state than an
in vitro model.
Ex vivo models are
frequently used for disease modeling, Drug Discovery and Development,
regenerative medicine, and tissue engineering. Ex vivo and in vitro
models represent an excellent alternative to animal testing.
There are three critical
differences between ex vivo and in vitro:
·
Ex vivo
approaches maintain a higher level of biological complexity by keeping the organ
or tissue structure intact, whereas in vitro experiments are typically
conducted with isolated cell(s).
·
In vitro
experiments offer more control over external variables and are less complex and
cost-effective, making them suitable for studying specific molecular or cellular
mechanisms. These characteristics make them indispensable for
high-throughput screening (HTS) of large libraries of
compounds to identify potential Drug candidates, even Drug repositioning.
Ex vivo experiments, while still controlled, introduce more variables
due to the complexity of tissue or organ systems.
·
Ex vivo
methods offer more physiologically relevant insights into the whole organism
than in vitro studies because they maintain some native interactions
with tissues or organs.
Note : Ex-vitro studies -
In plant tissue culture, for example, ex vitro refers to
the transition of plants or tissues from an artificial, sterile laboratory
environment to a natural or greenhouse environment.
Ex vivo in science refers to
experimentation done in or on tissues obtained from an organism and maintained
under optimum conditions mimicking the natural condition. The main advantage of
ex vivo models lies in the fact that there are controlled conditions at all
times, i.e., minimum alteration and variation in experimental models; secondly,
tests and measurements that were not possible to be conducted in living subjects
due to ethical issues could be easily carried out. The ex vivo model and
experimentation includes assays, measurement of physical, thermal, electrical,
mechanical, optical, and other tissue properties in varied conditions along with
determining treatment against cancerous tissues with the help of various imaging
techniques, etc.
Ex vivo models used for peptide studies mainly
involves intestinal studies conducted to either measure their bioavailability,
or secretion of mucus from goblet cells, or as simple as contraction of peptides
present in intestine. This may be since intestine is the first main organ from
where the basic metabolism, absorption, and distribution begins and thus is the
main site of action for all nutrients in general. Peptides, proteins, and dairy
hydrolysates alter the mucin secretion by changing the expression and number of
goblet cells and thus influence the dynamics of mucus, for example, casein
hydrolysates and β-casomorphin 7 increased mucin secretion in ex vivo
preparations of rat jejunum (Claustre et al., 2002; Trompette et al., 2003).
Another example is that of casoxin D (YVPFPPF), which showed opioid antagonistic
action in a field-stimulated guinea pig ileum preparation (ileum contraction).
The other most widely used area of ex vivo models
included role of peptide in cancer treatment and drug delivery. The studies have
been conducted using endothelial cells. Systemic delivery of macromolecular
proteins and peptide through pulmonary route could not be accessed accurately.
For this purpose ex vivo model of isolated perfused lung has been used in
various studies. It provided an accurate observation and separate analysis of
lung absorption and nonabsorptive clearance, i.e., phagocytosis and/or
metabolism, mucociliary clearance, etc. Such models have also been successfully
used for pulmonary insulin studies in isolated perfused rat lung.
Another research area where ex vivo models are
extensively exhausted involves assessment of phagocytosis and lymphocyte
proliferation response to determine immunomodulatory response. For phagocytotic
activity, macrophages isolated from peritoneal fluid are cocultured with
pathogen along with BAPs to observe the enhancement or suppression in their
activity as was seen in a study conducted in which β-casomorphin increased sheep
RBCs’ phagocytosis by murine peritoneal macrophages elucidating their role as
immunobooster. Thus ex vivo models play a pivotal role in research related to
efficacy studies in ways mentioned previously.
One widely performed ex
vivo study is the chickchorioallantoic membrane (CAM) assay. In this assay,
angiogenesis is promoted on the CAM membrane of a chicken embryo outside the
organism (chicken).
In addition to primary cell isolation
and cell lines, ex vivo models such as
freshly-dissected and cultured long bones have proven useful in the study of the
mechanical response of bone cells. For example, the activation of actin
contractions by load-induced Ca2 + signaling was confirmed through
axial compressive loading on an ex vivo tibia observed via fluorescent
microscopy. This system preserves the native bone environment, allowing both
cell level and tissue level factors to play into early biochemical responses.
Furthermore, this system can serve as a baseline method of analysis to address
numerous biological alterations made in transgenic mice, for example. Of note,
however, there are limitations in study duration of this ex vivo system since
cells cannot be kept alive indefinitely, and there is also of course the absence
of other organ systems influences (Fig.).
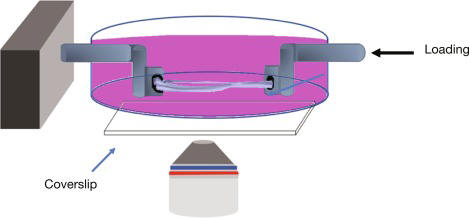
PURPOSE AND RATIONALE
Prolonged
administration of coronary drugs has been shown to increase the number and size
of interarterial collaterals of dogs and pigs after coronary occlusion. An
increased rate of development of collateral arteries was observed after physical
exercise in dogs, as well as after chronic administration of coronary dilating
drugs. An even more effective stimulus for collateral development is an acute or
gradual occlusion of one or several major coronary branches. Filling the
arterial coronary bed with a plastic provides the possibility to make the
collaterals visible and to quantify them.
PROCEDURE
Dogs
weighing 10–15 kg are anesthetized with pentobarbital sodium 30 mg/kg i.v. They
are respirated artificially and the thorax is opened. After opening of the
pericard, Ameroid cuffs are placed around major coronary branches. Gradual
swelling of the plastic material occludes the lumen within 3–4weeks. The dogs
are treated daily with the test drug or placebo. After 1 week recovery period
they are submitted to exercise on a treadmill ergometer. After 6weeks treatment,
the animals are sacrificed, the heart removed and the coronary bed flushed with
saline. The liquid plastic Araldite is used to fill the whole coronary tree from
the bulbus aortae. The aortic valves are glued together in order to prevent
filling of the left ventricle. Red colored Araldite is used to fill the arterial
tree. The venous part of the coronary vasculature can be filled with blue
colored Araldite from the venous sinus. The uniformity of the filling pressure,
the filling time, and the viscosity of the material are important.
Polymerization is complete after several hours. Then, the tissue is digested
with 35% potassium hydroxide. The method gives stable preparations which can be
preserved for a long time.
EVALUATION
Plastic
casts from drug treated animals are compared with casts from dogs submitted to
the same procedure without drug treatment.
CRITICAL ASSESSMENT OF THE METHOD
The
procedure allows impressive demonstration of the formation of arterial
collaterals. The results of post mortem Araldit impletion agree with the
functional results of experimental coronary occlusion.
Good laboratory practice (GLP) compliance in pharmacological
studies
GLP
comprises adherence to a set of formal, internationally agreed guidelines
established by regulatory authorities, aimed at ensuring the reliability of
results obtained in the laboratory. The rules (GLP Pocketbook,
1999; EEC directives 87/18/EEC, 88/320/EEC, available online:
pharmacos.eudra.org/F2/eudralex/vol-7/A/7AG4a.pdf) cover all stages of an
experimental study, from planning and experimental design to documentation,
reporting and archiving. They require, among other things, the assignment of
specific GLP-compliant laboratories, certification of staff training to agreed
standards, certified instrument calibration, written standard operating
procedures covering all parts of the work, specified standards of experimental
records, reports, notebooks and archives, and much else. Standards are
thoroughly and regularly monitored by an official inspectorate, which can halt
studies or require changes in laboratory practice if the standards are thought
not to be adequately enforced. Adherence to GLP standards carries a substantial
administrative overhead and increases both the time and cost of laboratory
studies, as well as limiting their flexibility. The regulations are designed
primarily to minimize the risk of errors in studies that relate to safety. They
are, therefore, not generally applied to pharmacological profiling as described
in this chapter. They are obligatory for toxicological studies that are required
in submissions for regulatory approval. Though not formally required for safety
pharmacology studies, most companies and contract research organizations choose
to do such work under GLP conditions.
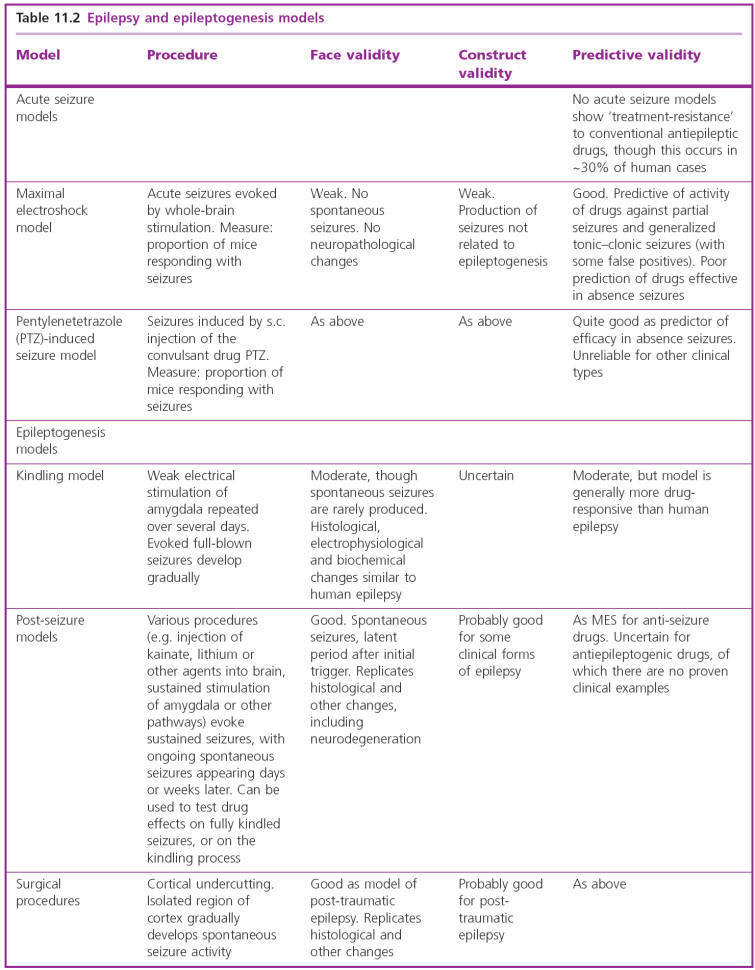
Lead Optimization
At this point, one or more
promising drug candidate hits have been identified and are ready to be promoted
to lead status. This entails verifying or optimizing the previously mentioned
qualities necessary for the drug candidate to be pursued: ease of synthesis,
adherence to the Lipinsky rules, target specificity, and efficacy against the
disease.
Lead optimization
Process
The lead
optimization (modification) process involves improvement of pharmacokinetic
(ADME) properties. The lead optimization process attempts to increase the
desired therapeutic potency (therapeutic index) and to decrease toxicity of the
parent molecule. The lead optimization process also offers specificity
optimization and molecular modification of agonist (lead) to antagonist and
vice versa. The lead optimization process involves the identification of the
pharmacophore and the auxophore. The functional features of molecules
responsible for the drug-receptor interaction and biological response is known
as a pharmacophore. The molecular features responsible for the conformational
change and molecular functions are considered as pharmacophoric part. The
remaining molecular features (atoms and groups) maintain the molecular integrity
and are called as auxophore. The removal of such groups abolishes the activity.
Similarly, the molecular features responsible for altered (increase or decrease)
potency refers to an auxophoric group.
Lead Optimization Approaches
Chemical
manipulation of molecules enhances their therapeutic function. The structural
geometry (size, shape and hydrogen bonding capacity) determines the structural
complementary and the receptor binding. The molecular groups are responsible for
the absorption, distribution and excretion pattern of the compound. Different
lead modification approaches widely applied are listed below.
1.
Functional group modification
2.
Structure-activity relationship
3. Molecular modification
4. Molecular hybridization
5. Bio-isosterism
6. Stereochemical aspects
1. FUNCTIONAL GROUP MODIFICATION
In general,
the polarity and ionization of compounds can be altered through functional group
modification. The antibacterial agent, carbutamide was found to have
anti-diabetic activity (side effect). Functional group modifications were
utilized in the improvement of the anti-diabetic potential and to remove the
antibacterial activity. The replacement of the amino group of carbutamide with
the methyl group retained the anti-diabetic function and removed antibacterial
potential. The molecule with methyl substitution is named as tolbutamide. The
change of methyl group with chloro group and shortening of liphophilic alkyl
side chain (butyl to propyl) generated another drug namely chlorpropamide. The
chlorpropamide has extended half life and six fold increase in activity.

2. STRUCTURE ACTIVITY RELATIONSHIP
The size and
shape of the molecules significantly contribute to biological activity. In most
instances the therapeutic effect of molecules depend on their structure. This
process involves collection and analysis of structural activity relationship
(SAR) data. The incorporation and replacement of functional groups generates
analogues with different size and shape, e.g., zolpidem.
The
structural modifications produce newer analogues with altered pharmacokinetic as
well as pharmacodynamic properties. Based on the structural dependence for the
intended biological functions, drugs are classified into structurally specific
and structurally non-specific drugs.
• Structurally
specific drugs: The activity and potency of structurally specific drugs are
very sensitive to even small change in chemical structure. The N-methyl
derivative of morphine (natural alkaloid) produces skeletal muscle relaxant
effect. Morphine has analgesic potential. The molecular function variation
suggests the importance of structure and receptor specificity.
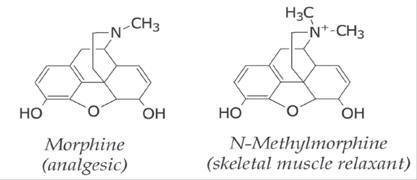
Structurally
non-specific drugs: The drugs such as gaseous anaesthetics do not depend on their
structures for their function. The activity of these molecules depends on their
physicochemical properties. The gaseous anaesthetics (halothane, isoflurane and
enflurane) produce their anaesthetic function by altering the membrane
electrochemical gradient sensing potential.
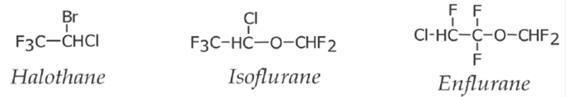
Structural Flexibility and Rigidity: The molecules having several
rotatable bonds adopt different conformations and influence the biological
activity. The introduction of rigidity or flexibility in the molecular structure
is the most common lead modification approach. The molecular conformation can be
locked by connecting certain atoms/groups (cyclization). The altered
conformation may enhance (agonist) or remove (antagonist) the biological
activity of parent molecule. The conformation responsible for the desired
pharmacological function is known as a bio-active conformation. The
oxo-burimamide, oxo-analogue of thioburimamide shows no anti-ulcer function. The
inter-molecular hydrogen bonding (rigidity) of oxoburimamide is reason for no
anti-ulcer activity. The thio group (exchange for oxo group of oxo-burimamide)
of thioburimamide would not establish hydrogen bond, hence elicits anti-ulcer
activity.

The exchange of N,N-dimethylamino groups (acyclic) of
chlorpheniramine into pyrrolidine (cyclic) group generated triprolidine. In this
case, the rigid (cyclic) as well as
flexible (acyclic) molecules exhibit anti-histaminergic functions. The
transformation of acyclic structural component into cyclic analogue may not
necessarily affect the molecular function.

3. MOLECULAR MODIFICATION
Homologation:
A process of increasing the molecular chain length of molecule by a constant
unit (e.g., methylene). The increase or decrease in the alkyl group size
(methylene shuffle strategy) influences the therapeutic potential. The molecular
modification either increases or decreases the biological activity of molecules.
The increase in carbon length in turn increases the molecular lipophilicity
(membrane penetration) and the bioavailability. This offers enhanced therapeutic
effect. The carbon chain length beyond certain level disturbs the optimal
balance between lipophilicity and hydrophilicity. The imbalance in molecular
properties decreases the bioavailability and therapeutic activity, (e.g., local
anaesthetics).
In the development of second generation sildenafil derivatives with increased
drug selectivity, the propyl group attached to the pyrazolo portion is exchanged
with ethyl group. Similarly sildenafil was modified into vardenafil.

Chain branching: The branched alkyl chains have more lipophilic character than
the corresponding straight alkyl chain (lower Log P). The chain branching also
interferes with bioactive conformation of the molecules and receptor binding.
The straight alkyl bridge in phenothiazine derivative of chlorpromazine produces
antipsychotic function. In contrast, promethazine with branched alkyl bridge
feature offers anti-histaminergic function.
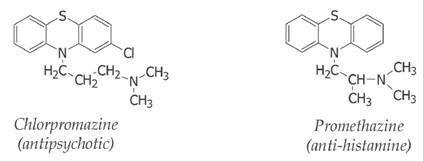
4. MOLECULAR HYBRIDIZATION
The
association of two individual molecules generate hybrid molecules (molecular
hybridization). In several cases, this improves pharmacokinetic and
pharmacodynamic functions compared with the individual effects of two drugs. For
example, the hybridization of aspirin and paracetamol produced acetaminosalol.
The hybrid molecules activate different targets and increase the therapeutic
index. The association of identical pharmacophoric entities (drug duplication,
triplication and tetraplication) produces more selective and potent therapeutic
effects. This process is known as molecular replication and the example is
di-aspirin.
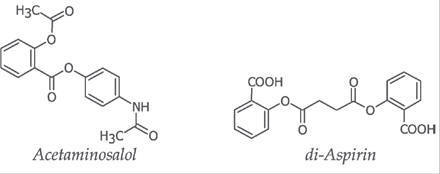
5. BIO-ISOSTERISM
Replacement
of atom or group of atoms in a molecule by another group with similar electronic
and steric properties is an important lead optimization process. Two molecules
or molecular fragments having identical number and arrangement of electrons are
termed as isosteres.
CO and N
2
…….CO = 6 + 8 = 14;
N2 = 7 +
7 = 14
These isosteres will have similar physicochemical properties in
most instances. It induces modifications in size, shape, electronic
distribution, chemical reactivity, liphophilicity and hydrogen bonding capacity
of molecules. The existence of such phenomenon is termed as isosterism. The
widespread application of the isosterism
concept to modify biological activity has given rise to the term
bio-isosterism. It is an important tool in rational drug design (RDD) process. •
Langmuir postulation (1919): The atoms or groups with the same number of
valence electrons are considered as isosteres and the phenomenon as isosterism.
• Grimm’s hydride displacement law (1925): Grimm hydride
displacement law states that the atoms belonging to group 4A, 5A, 6A and 7A in
the periodic table change their properties by adding a hydride. These hydride
added atoms are known as isoelectronic pseudo atoms. These pseudo atoms may
exhibit similar bio-physicochemic properties.
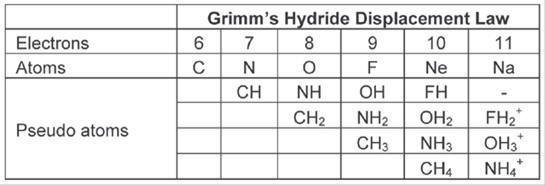
Erlenmeyer concept
(1932): Erlenmeyer
broadened and refined Grimm’s concept. He proposed isosteres as atoms, ions and
molecules with identical number of peripheral layer electrons. The elements in
the same column of the periodic table are isosteric.

Bio-isosterism is a special process of lead modification. Bio-isosteric
modification led to a fruitful yield of new and improved drug. The bio-isosteric
approach requires rigorous and careful analysis.
• Friedman concept (1951): Friedman defined bio-isosteres as compounds
similar to that of isosteres and having similar bioactivity.
• Thornber theory (1979): This theory states that the bio-isosteres are
subunits, groups and molecules possessing similar physicochemical as well as
bioactive properties.
• Alfred Burger hypothesis (1991): The compounds or groups possessing
near-equal molecular shape and volumes are considered as isosteres. The similar
electronic distribution offers similar physicochemical properties to the
isosteres.
Classification
1.
Classic bio-isosteres:
The bio-isosteric groups are classified based on their electronic features and
valence electrons. (a) Monovalent groups (b) Divalent groups (c) Trivalent
groups (d) Tetravalent groups (e) Ring equivalents
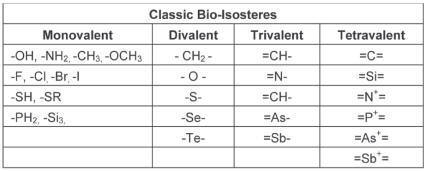
2.
Non-classic bio-isosteres
(a) Cyclic vs non-cyclic (b) Functional groups (c) Retroisomerism
CLASSIC BIO-ISOSTERISM
Modification of Monovalent Groups
Hydrogen bonding modification: The
similar steric size, spatial arrangement and the hydrogen bond donor/acceptor
nature is utilized in the lead modification process.
•
Fluorine-hydrogen interchange: Hydrogen and fluorine are sterically
similar, but differ in their electronegativity. 5-Fluorouracil (anticancer drug)
and uracil (natural substrate for thymidylate synthase) is an example. In
5fluorouracil, the fluoro atom in the fifth position of uracil (in place of
hydrogen in uracil) is responsible for the altered thymidylate synthase pathway.
The negative inductive effect of fluorine atom, inhibits the formation of
thymidine (thymidylate synthase product). This property offers
anti-proliferative (anticancer) function to 5-fluorouracil.

Amino-hydroxyl interchange: The
replacement of hydroxyl group of folic acid with amino group generates
aminopterin (anti-bacterial). Tautomeric nature and hydrogen bonding capability
of amino group of aminopterin (compared with hydroxyl group) facilitates the
bonding with dihydrofolate reductase. This enzyme is essential for the cell
growth function. Thus aminopterin competes with folic acid and inhibits enzyme
binding. This enzyme inhibition in turn inhibits the folic acid metabolism and
offers antibacterial activity.

Thiol-hydroxyl interchange: In
6-thioguanine (anticancer drug) the keto group of the guanine is exchanged with
thio group. This change contributes to cell proliferation inhibition
(anticancer).
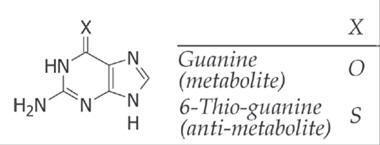
•
Grimm’s concept: The soft metabolic site (methyl group; -CH3) present in
SC-58125 reduces their systemic concentration (reduced activity). Hence, the
bio-isosteric approach of methyl (-CH3) group replacement with bioisosteric
amino (-NH2) group generated celecoxib (selective and potent cyclooxygenase-II
inhibitor).
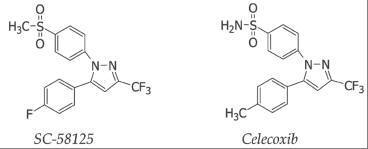
Modification of Divalent Groups The divalent bio-isosteres include methylene (-CH2-), secondary
amino (-NH-), ether (-O-) and thioether (-S-) functionalities. It also includes
-C=N-, -C=O and -C=S. In all these groups, the bond angle and corresponding
molecular conformations are nearly equal. These divalent bio-isosteres were
utilized for eliminating the pharmacokinetic difficulties with retention of
biological activity.
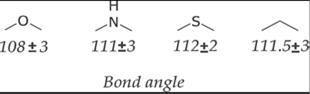
The
bioisosteric replacement of divalent ether (-O-) group of procaine (local
anesthetic) with secondary amine group (-NH-) generated procainamide
(antiarrthythmic).
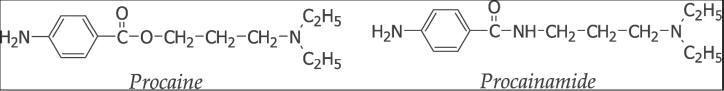
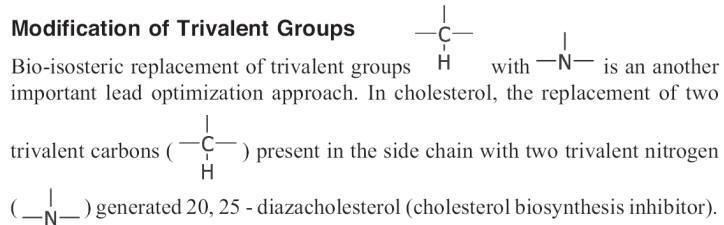
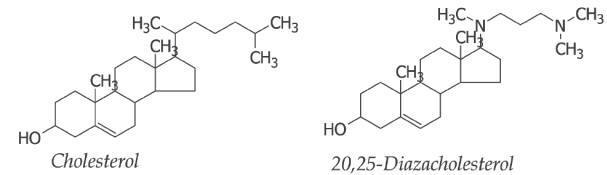
Modification of Tetravalent Groups
Replacement
of quaternary nitrogen with tertiary carbon is classical example of this type.
Carnitine acyl transferase inhibitors include replacement of the hydroxyl group
of carnitine with amino and as well as quarternary nitrogen with carbon to get
trimethyl ammonium group.
Ring Equivalents
Benzene and
thiophene shares similar physicochemical properties and are known as ring
equivalents. This inspired medicinal chemists to apply this concept in drug
design.
Pyrazole-isoxazole-pyridine exchange: The replacement of pyrazole of celecoxib with isoxazole and
pyridine produced more potent cyclooxygense-II inhibitors namely valdecoxib and
etoricoxib.
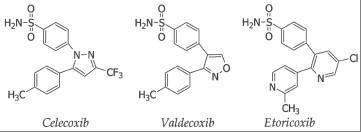
Non-Classical Bio-Isosterism
•
Cyclic vs non-cyclic: Local anaesthetic agents lidocaine and mepivacaine
are best examples of this kind. The open chain lidocaine and closed ring
structure in mepivacaine do not change their biological activity.

Functional groups:
Diverse functional groups are known for their bioisosteric relationship. The
angiotensin-II antagonist losartan was developed by exchanging carboxylic group
of EXP 7711 (lead) with tetrazole group.
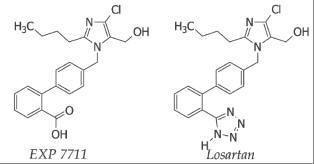
The exchange of keto group (-C=O) group of oxo-tolrestat with thio (-C=S) group
generated more potent aldose reductase inhibitor namely tolrestat. The
oxotolrestat is less active orally.
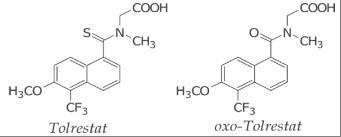
·
Retroisomerism:
It involves the inversion of functional groups. The inversion of amide group of
procainamide into anilide group generated more potent local anesthetic drug
lidocaine.
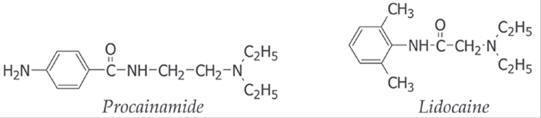
6. STEREOCHEMICAL ASPECTS
The relative
functional difference (defined structural features) of active molecules provided
insight about the receptor. The spatial arrangement of atoms determines the
receptor affinity and pharmacological response. The biological receptors (made
up of L-amino acids) also exhibit chiral (steric) features and offers specific
affinities for the binding. These chiral molecules are optically active
molecules (stereoisomers) and include enantiomers, diasteroisomers and geometric
isomers. The chiral nature of receptor demands for the chiral specific drugs.
Chirality:
An atom attached with four different atoms or groups (substituents) bears
asymmetric center (chiral center). The molecules with chiral centers are called
as chiral compounds.

Enantiomers: The optical active isomer which rotate plane polarized light in
right direction (clock wise) is known as a dextro-rotatory (d-isomer) and
left direction (anti-clock wise) is known as a levo-rotatory (l-isomer).
These stereoisomers are called as enantiomers, which show altered affinity for
receptors, different binding energies and chemical properties. In case of
chlorpheniramine (antihistamine), the dextro isomer is more potent than the levo
isomer.
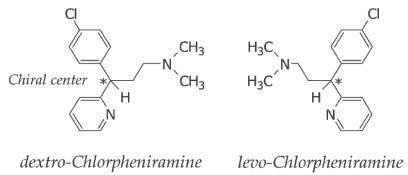
·
Eutomer:
An active and potent enantiomer is termed as eutomer. e.g.,
dextrochlorpheniramine.
•
Distomer: A less active enantiomer is termed as distomer. e.g.,
levochlorpheniramine. Eutomer for one pharmacological effect may be distomer for
another pharmacological effect. The dextro isomer of propoxyphen
(dextropropoxyphen) is analgesic and levo-propoxyphen is anti-tussive. In terms
of analgesic function dextro-propoxyphen is eutomer and levopropoxyphen is
distomer. Levopropoxyphen is eutomer for anti-tussive function.
• Hybrid
drug: An enantiomer having different pharmacological potential through
different molecular mechanism are termed as hybrid drug. An S(+) enantiomer of
ketoprofen is non-steroidal anti-inflammatory agent, where as R(-) enantiomer is
useful in the treatment of periodontal disease.
•
Isomeric ballast: The distomer with undesirable effects (toxicity) are
termed as isomeric ballast. The S-isomeric form thalidomide (hypnotic and
sedative) produces teratogenic effects and is good example for isomeric ballast.

Diastereomers:
The non-super imposable stereoisomers with more than one chiral center are
called as diastereomers. The b-receptor antagonistic drug labetalol is an
example. The R,R isomeric form of the drug shows selective b -receptor blockade
effect, but the S,R isoform produce á-receptor blocking effect.
Racemic switch: The introduction of single enantiomeric form of the racemate
drug is called as racemic switch (chiral switch). The single enantiomer drugs
esomeprazole (S-omeprazole; antiulcer), levocetrizine (R-cetrizine;
antihistamine) and levalbuterol (R-salbutamol; anti-asthmatic) are the examples.
Geometrical isomers: The restricted rotation around the double bond produce
geometrical isomers (cis and trans; Z and E). The anti-histaminergic drug
triprolidine and a synthetic analogue of oestrogen diethylstilbestrol are
examples. Triprolidine is active only in Z-configuration (cis, H/Pyridyl). The
E-isomeric form of diethylstibestrol produce required response.
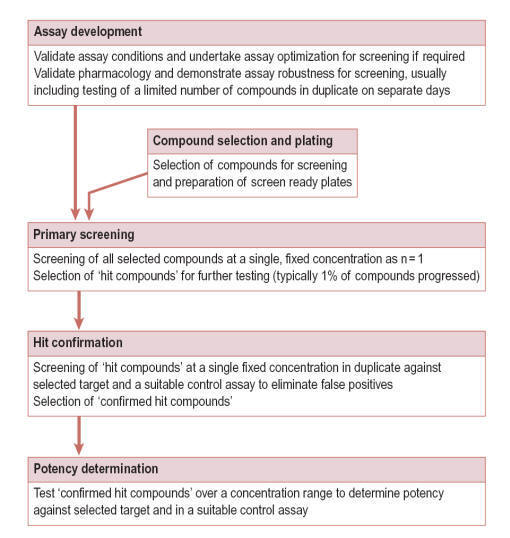

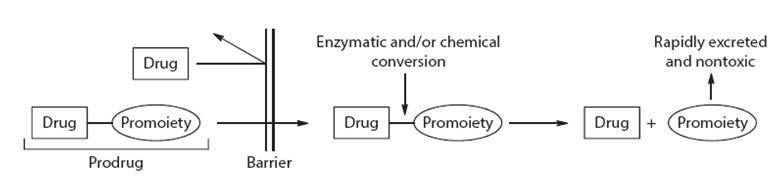
Relative to in vivo ECG recording,
ex vivo ECG procedures are very
straight-forward; requiring no anesthetics, paralytics or external perfusion. In
the absence of pericardial tissue, AgCl wire electrodes can be positioned in
very close proximity to the heart and the ECG signal is easily measured using
even rudimentary amplifiers (Lin et al. 2015), as illustrated in Fig. 3B. An
obvious limitation of the ex vivo ECG is excision of the heart, which can be
technically challenging. However, there are surgical procedures for rapid
zebrafish heart isolation (Arnaout et al. 2014)(Lin et al. 2015). By approaching
the heart from the cranial aspect or from the caudal aspect, these techniques
limit the possibility of tissue damage caused by ventral incisions and can be
completed in <5 min with minimal training.