DRUG DESIGNING
The shortcoming of traditional drug discovery; as well as the allure of a more deterministic approach to combating disease has led to the concept of "Rational drug design" (Kuntz 1992). Nobody could design a drug before knowing more about the disease or infectious process than past. For "rational" design, the first necessary step is the identification of a molecular target critical to a disease process or an infectious pathogen. Then the important prerequisite of "drug design" is the determination of the molecular structure of target, which makes sense of the word "rational".
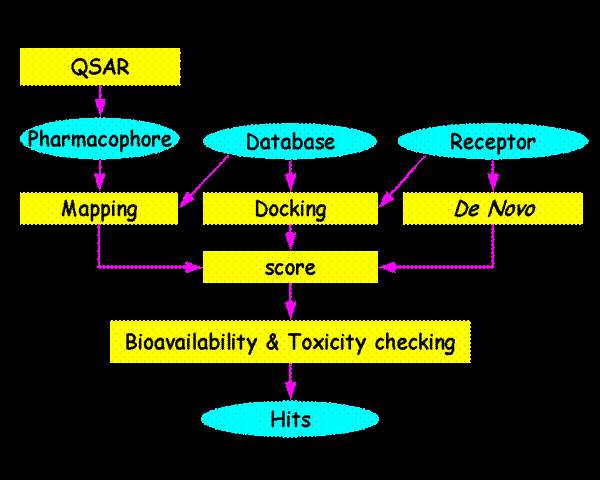
In fact, the validity of "rational" or "structure-based"drug discovery rests largely on a high-resolution target structure of sufficient molecule detail to allow selectivity in the screening of compounds. Simple flowchart for drug designing shown in the figure:
Drug designing basically of two types namely ligand based approach or receptor based approach. In both the case the point of centre only differ but requirement of receptor and ligand essential in both the case. By considering these facts, the following steps and online tools shown below for drug designing.
Drug designing steps were usually divided into steps as follows:
I. LIGAND PREPARATION
II. RECEPTOR PREPARATION
III. DOCKING
IV. BINDING AFFINITY STUDIES
1. LIGAND PREPARATION:
Ligand preparation further divided into different heading namely, ligand retrieval or collection, liand conversion and ligand analysis
a. Ligands Collection:
1. DRUGBANK:http://www.drugbank.ca/
The DrugBank database is a unique bioinformatics and cheminformatics resource that combines detailed drug (i.e. chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e. sequence, structure, and pathway) information. The database contains nearly 4800 drug entries including >1,480 FDA-approved small molecule drugs, 128 FDA-approved biotech (protein/peptide) drugs, 71 nutraceuticals and >3,200 experimental drugs. Additionally, more than 2,500 non-redundant protein (i.e. drug target) sequences are linked to these FDA approved drug entries. Each DrugCard entry contains more than 100 data fields with half of the information being devoted to drug/chemical data and the other half devoted to drug target or protein data.
2. PUBCHEM: http://pubchem.ncbi.nlm.nih.gov/
PubChem provides information on the biological activities of small molecules. It is a component of NIH's Molecular Libraries Roadmap Initiative.
3. CHEMBANK : http://chembank.broad.harvard.edu/welcome.htm
It contains part of an electronic structure collection donated by Tudor Oprea. This set originates from a compilation of ~4.5 million compounds commercially available in August 2002. These were collected from CDs offered by 10 vendors. The structures were processed into a standardized format using OpenEye’s FILTER software (http://www.eyesopen.com/products/applications/filter.html). Compliance with Lipinski’s Rule-of-5 was enforced (no violations allowed), and several "undesirable" chemical substructures were removed. A low-value for drug-like scores (scores > 0.2) was implemented in order to further remove chemicals that were very different from the then-accepted medicinal chemistry space. Approximately ~2.5 million compounds passed these filters, and these were subsequently subjected to diversity selection using D-optimal design and a 2D-based descriptor system (mostly topological indices, atom counts, and LogP-type descriptors), in order to realize the final collection of ~800K compound structures.
4. LIGAND EXPO: http://ligand-expo.rcsb.org/ld-search.html
Ligand Expo (formerly Ligand Depot) provides chemical and structural information about small molecules within the structure entries of the Protein Data Bank. Tools are provided to search the PDB dictionary for chemical components, to identify structure entries containing particular small molecules, and to download the 3D structures of the small molecule components in the PDB entry. A sketch tool is also provided for building new chemical definitions from reported PDB chemical components.
5. Small molecules search by descriptors: http://www.scfbio-iitd.res.in/software/nrdbsm/drugsearch.jsp
NRDBSM database is aimed specifically at virtual high throughput screening of small molecules and their further optimization into successful lead-like candidates. The NRDBSM besides facilitating focused searches in larger databases once a hit is identified should also help in finding a small number of hits for further optimization. A Search engine is available for querying NRDBSM based on the properties mentioned.
b. Ligand conversion (format conversion):
1. Smileconvertor - Cactus: http://cactus.nci.nih.gov/services/translate/
This tool was used to convert smiles into PDB, SDF, Mol formats and both in 2D and 3D formats.
2. 2Dto3D convertor: http://www.molsoft.com/2dto3d.html
ICM 2D to 3D converter convertion functionality allows construction of icm molecular objects from smiles-strings and creates optimized 3D structures using MMFF atom type assignment and force-field optimization. You can use this page to test convert your chemical structure in smiles-format to 3D and view it using our Java applet-based viewer. A simplified 3-D graphical object will be created at the Molsoft server using the ICM software, and your browser won't have to download too much data.
3. CORNIA: http://www.molecular-networks.com/online_demos/corina_demo
This tool was used to generate 3D structures in SDF format.
4. PRODRG: http://davapc1.bioch.dundee.ac.uk/prodrg/index.html
This WWW PRODRG server will convert coordinates for small molecules in PDB format to the following topology formats: GROMOS, GROMACS, WHAT IF, REFMAC5, CNS, O, SHELX, HEX and MOL2. In addition, coordinates for hydrogen atoms are generated. You can now also sketch your small molecule in a simple text editor, and paste this into the window below. You will be returned all of the above topology files + a GROMOS energy
c. Ligand Analysis:
i)Molecular Descriptors:
Molecular descriptors plays crucial role in the drug identification area. So it is essential to know and have the molecular descriptors values regarding ligands. Molecular descriptors predicted through QSAR and QPAR models.
1. MOLINSPIRATION: http://www.molinspiration.com/cgi-bin/properties
Draw your molecule and press the [Calculate Properties] or [Predict Bioactivity] button. When the JME input is not working on your computer, try our NEW WebME Ajax editor NEW or paste a raw SMILES here. You may wish to check also our Property Prediction FAQ or more information about calculated properties and drug likeness.
2. EDRAGAON: http://www.vcclab.org/lab/edragon/start.html
E-DRAGON is the electronic remote version of the well known software DRAGON, which is an application for the calculation of molecular descriptors developed by the Milano Chemometrics and QSAR Research Group of Prof. R. Todeschini. These descriptors can be used to evaluate molecular structure-activity or structure-property relationships, as well as for similarity analysis and highthroughput screening of molecule databases. DRAGON provides more than 1,600 molecular descriptors that are divided into 20 logical blocks. The user can calculate not only the simplest atom type, functional group and fragment counts, but also several topological and geometrical descriptors. The first release of DRAGON dates back to 1997. Updates and inclusions of new molecular descriptors are regularly made in order to advance research in QSAR.
3. MOLEDB: http://michem.disat.unimib.it/mole_db/
Molecular descriptors data base was used to get different molecular descriptors for different ligands and drugs which already stored in databases.
4. Model: http://jing.cz3.nus.edu.sg/cgi-bin/model/model.cgi
MODEL - Molecular Descriptor Lab for Computing structural and physichemical properties of molecules from their 3D structures.
5. Lipinski’s rule prediction: http://www.scfbio-iitd.res.in/software/drugdesign/lipinski.jsp
Lipinski rule of 5 helps in distinguishing between drug like and non drug like molecules. It predicts high probability of success or failure due to drug likeness for molecules complying with 2 or more of the following rules:
ii) ADME Prediction and Druglikliness prediction:
The success of an drug maily depends upons its ability to enter into the host and not producing any adverse effect on it. These properties were tested by ADME (Absorption, Distribution and Metabolism and Excretion) prediction tools and lead and druglikliness of the chemicals also determined in this phase of designing.
1. ADMETools: http://www.simcyp.com/ProductServices/FreeADMETools/
2. ADME DATABASE: http://modem.ucsd.edu/adme/databases/databases_extend.htm
3. Drug Likliness: http://www.molsoft.com/mprop/
II. RECEPTOR PREPARATION:
For drug to act, target is necessary which might be of receptor or enzyme or hormone type. Initially, the receptor should be prepared by structure modeling method or it might be download from the structure databases. After modeling or downloading from corresponding sources, it should be prepared properly for docking which might be achieved by using binding site analysis tools and target determination tools.
i) Bindingsite Analysis:
1. CASTp: http://cast.engr.uic.edu/castp/index.php
2. Protein Pocket: http://sts.bioe.uic.edu/pni/
3. Protein Cavities: https://honiglab.c2b2.columbia.edu/MarkUs/cgi-bin/submit.pl
4. MEDOCK: http://bioinfo.mc.ntu.edu.tw/medock/step1.html
The MEDock (Maximum-Entropy based Docking) web server is aimed at providing an efficient utility for prediction of ligand binding site. A major distinction in the design of MEDock is that its global search mechanism is based on a novel optimization algorithm that exploits the maximum entropy property of the Gaussian distribution.
5. ODA : http://www.molsoft.com/cgi-bin/oda.cgi
ODA (Optimal Docking Areas) is a new method to predict protein-protein interaction sites on protein surfaces. It identifies optimal surface patches with the lowest docking desolvation energy values as calculated by atomic solvation parameters (ASP) derived from octanol/water transfer experiments and adjusted for protein-protein docking. The predictor has been benchmarked on 66 non-homologous unbound structures, and the identified interactions points (top 10 ODA hot-spots) are correctly located in 70% of the cases (80% if we disregard NMR structures).
ii) Target Determination:
1. TarFisDock : http://www.dddc.ac.cn/tarfisdock/index.php
TarFisDock is a web server for identifying drug targets with docking approach. Given a small molecule which can be drug, drug candidate, natural product, or new synthetic compound, TarFisDock docks it into the protein targets in PDTD (Potential Drug Target Database), and outputs the top 2%, 5% or 10% candidates ranked by the energy score, including their binding conformations and a table of the related target information. The server is freely accessible for anonymous user. And one user's result is protected from being retrieved by another. However users are encouraged to fill in a very simple registration form for better safety and convenience. Now submit your molecular structure(in mol2 format) by clicking
2. TTD: http://bidd.nus.edu.sg/group/cjttd/ttd.asp
A database to provide information about the known and explored therapeutic protein and nucleic acid targets, the targeted disease, pathway information and the corresponding drugs/ligands directed at each of these targets. Also included in this database are links to relevant databases that contain information about the function, sequence, 3D structure, ligand binding properties, enzyme nomenclature and related literatures of each target. This database currently contains 1535 targets and 2107 drugs/ligands.
3. Supertarget Search: http://bioinf-apache.charite.de/supertarget_v2/index.php?site=home
4. Binding Target Determination: http://www.bindingdb.org/bind/vsOverview.jsp
III. DOCKING:
After collecting, preparing ligands and receptors, they should be assessed for their interaction ability with docking procedure. There were many docking tools are available online. Docking studied under two heads namely, protein-ligand docking and protein-protein docking.
i) Protein-Ligand Docking
1. Patch dock: http://bioinfo3d.cs.tau.ac.il/PatchDock/
2. ParDock: http://www.scfbio-iitd.res.in/dock/pardock.jsp
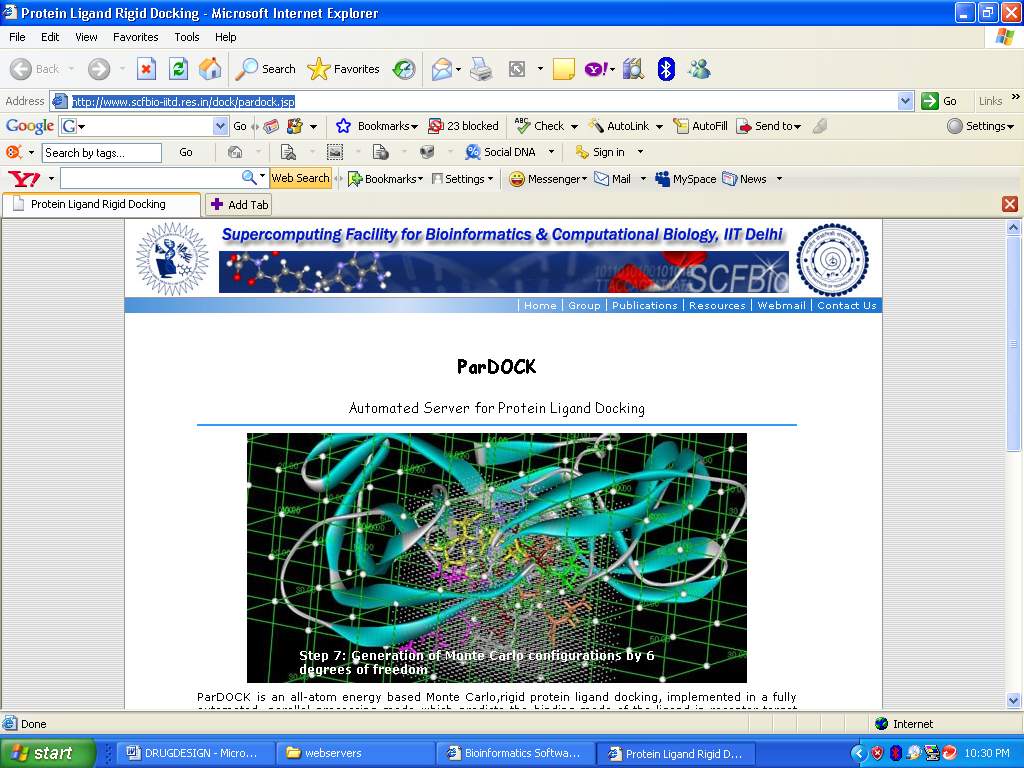
ParDOCK is an all-atom energy based Monte Carlo,rigid protein ligand docking, implemented in a fully automated, parallel processing mode which predicts the binding mode of the ligand in receptor target site.
ii) Protein-Protein Docking:
1. ROSIE: http://rosettadock.graylab.jhu.edu/docking2/submit
2.FIREDOCK: http://bioinfo3d.cs.tau.ac.il/FireDock/
3. CLUSPRO: http://cluspro.bu.edu/~rb/cluspro/login/main.php
Research in the Structural Bioinformatics lab, headed by Sandor Vajda, focuses on the recognition of proteins and small molecules by protein receptors. Studying protein-protein interactions is crucial for a better understanding of processes such as metabolic control, signal transduction, and gene regulation, whereas the ability to dock small ligands to proteins is the key to rational drug and vaccine design strategies. Both problems become much more difficult if no x-ray structure of the protein is available. Accordingly, our main research areas are (1) the development of efficient protein docking algorithms, (2) docking of small ligands to proteins, primarily for the characterization of binding sites, and (3) homology modeling of proteins.
4. Vakser Lab: http://vakser.bioinformatics.ku.edu/resources/gramm/grammx/
This is the Web interface to our current protein docking software made available to the public. This software is different from the original GRAMM, except that both packages use FFT for the global search of the best rigid body conformations.
5. 3DGarden: http://www.sbg.bio.ic.ac.uk/3dgarden/index.cgi
3DGarden is an integrated software suite for performing protein-protein docking. For any pair of protein structures specified by the user, 3DGarden's primary function is to generate an ensemble of putative complexed structures and rank them. The highest-ranking candidates constitute predictions for the structure of the complex. 3DGarden cannot be used to decide whether or not a particular pair of proteins interacts. 3DGarden cannot currently be used for docking DNA/RNA structures with proteins or with other DNA/RNA.
IV. BINDING AFFINITY STUDIES:
1. DRUGSCORE: http://pc1664.pharmazie.uni-marburg.de/drugscore/
DrugScoreONLINE is a web-based user interface for the knowledge-based scoring functions DrugScoreCSD and DrugScorePDB. DrugScoreONLINE enables you to score protein-ligand complexes of your interest and to visualize the per-atom score contributions as illustrated in the figures shown below. Blue spheres denote favorable interactions whereas red spheres stand for disfavorable ones. The sizes of the spheres correlate with the contributing per-atom scores.
2. BAPPL : http://www.scfbio-iitd.res.in/software/drugdesign/bappl.jsp
Binding Affinity Prediction of Protein-Ligand (BAPPL) server computes the binding free energy of a non-metallo protein-ligand complex using an all atom energy based empirical scoring function.
3.AFFINITY DB: http://pc1664.pharmazie.uni-marburg.de/affinity
AffinDB is a database of affinity data for structurally resolved protein–ligand complexes from the Protein Data Bank (PDB). It is freely accessible at http://www.agklebe.de/affinity. Affinity data are collected from the scientific literature, both from primary sources describing the original experimental work of affinity determination and from secondary references which report affinity values determined by others. AffinDB currently contains over 730 affinity entries covering more than 450 different protein–ligand complexes. Besides the affinity value, PDB summary information and additional data are provided, including the experimental conditions of the affinity measurement (if available in the corresponding reference); 2D drawing, SMILES code and molecular weight of the ligand; links to other databases, and bibliographic information. AffinDB can be queried by PDB code or by any combination of affinity range, temperature and pH value of the measurement, ligand molecular weight, and publication data (author, journal and year). Search results can be saved as tabular reports in text files. The database is supposed to be a valuable resource for researchers interested in biomolecular recognition and the development of tools for correlating structural data with affinities, as needed, for example, in structure-based drug design.
4. LIGAND-PROTEIN DB: http://lpdb.chem.lsa.umich.edu/
In computational structure-based drug design, the scoring functions are the cornerstones to the success of design/discovery. Many approaches have been explored to improve their reliability and accuracy, leading to three families of scoring functions: force-field-based, knowledge-based, and empirical. The last family is the most widely used in association with docking algorithms because of its speed, even though such empirical scoring functions produce far too many false positives to be fully reliable. In this work, we describe a World Wide Web accessible database that gathers the structural information from known complexes of the PDB with experimental binding data. This database, the Ligand-Protein DataBase (LPDB), is designed to allow the selection of complexes based on various properties of receptors and ligands for the design and parametrization of new scoring functions or to assess and improve existing ones. Moreover, for each complex, a continuum of ligand positions ranging from the crystallographic position to points on the surface of the protein receptor allows an assessment of the energetic behavior of particular scoring functions.
5. PEARLS: http://ang.cz3.nus.edu.sg/cgi-bin/prog/rune.pl
OTHER SITES
DENOVO DOCKING-GWIDD http://gwidd.bioinformatics.ku.edu/home
GWIDD is a comprehensive resource for genome-wide structural modeling of protein-protein interactions. It contains interaction information for multiple organisms. The structures of the participating proteins are modeled or crystallographic coordinates are retrieved, if available, and docked by GRAMM-X. The resource is not restricted to interactions in the GWIDD database - other sequences or structures may be entered at various stages.
CLINICAL TRIALS: http://www.centerwatch.com/
Drug Designing Supersite: http://www.drugdesign.org/DrugDesignWebEntries/index.html
Complete docking suite : http://www.scfbio-iitd.res.in/sanjeevini/sanjeevini.jsp
OSDD bioinformatics link : http://www.osddbengaluru.net/facilities.php?option=Bioinformatics
Dogsitescorer : http://dogsite.zbh.uni-hamburg.de/
Bioinformatics tool : http://www.vetbifguwahati.ernet.in/tools.html
Bioinformatics course online - https://cbbioinfo.appspot.com/cbbioinfo/