
DNA SEQUENCING
Molecular cloning allows the isolation of individual fragments of DNA in quantities suitable for detailed characterization, including the determination of nucleotide sequence. Indeed, determination of the nucleotide sequences of many genes has elucidated not only the structure of their protein products, but also the properties of DNA sequences that regulate gene expression. Furthermore, the coding sequences of novel genes are frequently related to those previously studied genes, and the functions of newly isolated genes can often be correctly deduced on the basis of such sequences similarities.
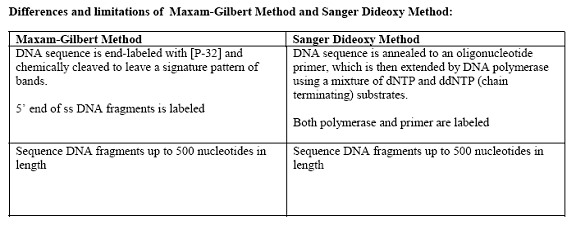
There are mainly five methods available for DNA sequencing, namely, Chemical method, Enzymatic method, Automated sequencing, shotgun method and cycle sequencing.
1. Maxam-Gilbert Method (Chemical
or Cleavage Method):
In this method, initially 5’-end of the DNA strand is labeled with 32P using enzymes like Alkaline Phosphatase and Polynucleotide Kinase which are used to remove phosphate group and add phosphate group at 5’-end respectively. The phosphate group donor was 32P labeled [g-32P] ATP. The other radioactive materials like 33P and 35S also used for labeling for sharper image and greater lifetime respectively when compared to 32P. The radioactive DNA is then denatured with NaOH and the two single strands are separated by electrophoresis. The sequencing procedure is as follows:
A sample containing the purified single strands is divided into two portions. To one portion (1), dimethyl sulphate is added which methylates purines; however, G is methylated five times more effectively than A. An important feature is that the reaction is stopped before completion. It is allowed to proceed to the extent that about one purine per single strand is methylated. Methylation occurs at random places and so that the particular A or G that is methylated differs in each strand. The methylated same is divided into two portions, 1a and 1b.
Sample 1a is heated. This treatment removes all methylated bases leaving the deoxyribose. The sample is then treated with alkali. This breaks the sugar-phosphate chain at the place of the base that has been removed. This heating treatment produces a set of fragments of varying size and the differing numbers of nucleotides in each fragment is determined by the different positions of the methylated G or A. Because G is methylated more often than A, sample 1a has the G-only fragments. Sample 1b is not heated but treated with dilute acids, which removes methylated A and some G. Then it is treated with alkali to break the sugar phosphate chain at the place where an A was removed. Therefore, in sample 1b, fragments are produced whose size is determined mainly by the position of the methylated A and also some G. These are called A + G fragments. Note that every G-only fragment size is also present in the A + G sample. Two samples 1a and 1b are now electrophoresed in agarose containing urea, a denaturant which prevents hydrogen bonding and thus keeps the fragments single stranded. After electrophoresis, the bonds are located by autoradiography. The single terminal 32P atom
which was added before the methylation reaction is the sole source of radioactivity. When a 5’-32P-labelled molecule is cleaved, only one of the two fragments contain 32P and only that one is detected. The position of A and G in the single strand are determined by the following rules. They are
Sample 2 is used to identify the positions of C and T. This sample is also divided into two portions 2a and 2b. One sample (2a) is treated with hydrazine and the other (2b) with hydrazine + NaCl. Hydrazine reacts with C and T but not with A or G whereas hydrazine + NaCl reacts with C only. This is followed by treatment with piperidine. This breaks the sugar-phosphate backbone at the 5’side of each base that has reacted with hydrazine. The sizes of the cleaved fragments are determined by the positions of both C and T and by C only in the gel. Thus, after electrophoresis and autoradiography, the positions of C and T are determined by the following rules. They are
All four samples (1a, 1b, 2a & 2b) are electrophoresed simultaneously so that all bands are seen in a single gel. Thus, the sequence can be read off directly from the gel. The 5’-end of the strand sequence present at the bottom of the gel and vice versa. One of the main disadvantages of this method is that it is not possible to identify the first base of the single strand. But second strand sequencing overcomes this problem and also it might be used for confirmation of the sequence of the first strand.
2. Sanger Method (Enzymatic or Termination Method):
The most common method of DNA sequencing is based on premature termination of DNA synthesis resulting from the inclusion of chain termination dideoxynucleotides in DNA polymerization reaction.
Principle:
For this method, specific terminators of DNA chain elongation 2’,3’-dideoxynucleoside triphosphates were synthesized. These molecules can be incorporated normally into a growing DNA chain through their 5’- triphosphates groups. However, they cannot form phosphodiester bonds with the next incoming deoxynucleotide triphosphates (dNTPs). When a small amount of a specific dideoxy NTP is included along with the four deoxyNTPs normally required in the reaction mixture for DNA synthesis by DNA polymerase, the products are a series of chains that are specifically terminated at dideoxy residue. This forms the basis for Sanger’s method.
Procedure:
Initially single strand DNA prepared through denaturation process. Then single strand DNA was mixed with a short end labeled piece of DNA (Primer) that is complementary to the end of single strand DNA. Labeling of primer carried out using enzymes like Alkaline Phosphatase and Polynucleotide Kinase. After primer annealed to DNA, Sample was divided into four portions in four tubes. In each tube, along with DNA, Primer, DNA polymerase, a carefully controlled ratio of one particular dideoxynucleotide with its normal deoxynucleotide, and the other three dNTPs added.
In each tube, DNA polymerase polymerizes normally from primer by utilizing nucleotides. When ddNTP is incorporated, the growth of that chain will stop. If the correct ratio of ddNTP: dNTP is chosen, a series of labeled strands will result, the lengths of which are dependent on the location of a particular base relative to the end of the DNA.
After suitable time period, the resultant labeled fragments in each tube separated by size on an acrylamide gel. The separated fragments were detected by exposure of the gel to x-ray film through the process of autoradiography. From the band developed in each lane of the autoradiograph and knowledge of which lane contain which base, the sequence of the complementary sequence can be obtained. From the complementary sequence, the sequence of the original strand can be easily determined with the help of Watson and crick base pairing rule. Thus Sanger method is used for DNA sequencing.

Normal DNA synthesis by dNTPs:

DNA synthesis blocked by ddNTP:


3. Automated Sequencing:
Large scale DNA sequencing is frequently performed using automated systems, which use fluorescent-labeled primers or fluorescent-labeled dideoxynucleotides in dideoxynucleotide sequencing reactions. As the newly synthesized DNA strands are electrophoresed through a gel, they pass through a laser beam that excites the fluorescent label. The resulting emitted light is then detected by a photomultiplier and a computer collects and analyzes the data. This type of automated DNA sequencing has enabled the large-scale analysis required for determination of the complete sequence of the human genome, as well as the genome sequences of a number of species of bacteria, yeast, Arabidopsis, C. elegans, Drosophila and the mouse.
This was achieved in three ways namely Four Reaction / Four Gel Systems, Four Reaction / One Gel Systems and One Reaction / One Gel Systems.
A. Four Reaction / Four Gel Systems:
The primer is linked at its 5’-end to a highly fluorescent dye and the chain extension reactions are carried out in four separate vessels using Sanger’s principle. The reactions products are then subject to sequencing gel electrophoresis in four parallel lanes and the order in which the fluorescent fragments pass through the gel is recorded by a laser activated fluorescent detection system. Four gels were used because single fluorescent dye used as label.

B. Four Reaction / One Gel Systems:
The primers used in each of the four chain extension reactions are each 5’-end linked to a differently fluorescing dye. The separately reacted mixtures are combined, subjected to sequencing gel electrophoresis in a single lane and the terminal base on each fragment identified according to its characteristic fluorescence spectrum. Still four lanes were utilized because primer is labeled.


C. One Reaction / One Gel Systems:
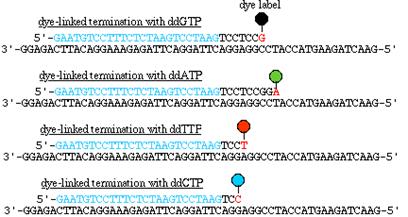

Each of the four ddNTPs used to terminate chain extension is covalently linked to a differently fluorescing dye, the chain-extension reaction is carried out in a single vessel, the resulting fragment mixture is subjected to sequencing gel electrophoresis in a single lane and the terminal base on each fragment is identified according to its characteristic fluorescence spectrum.
The fluorescence output is stored in the form of chromatograms:

This system can identify approximately 10,000 bases per day, in contrast to approximately 50,000 bases per year that a skilled operator can identify using the manual methods like chemical cleavage and enzymatic methods. Sequencing rates have been further increased by automating the setup of DNA sequencing reactions through the use of robotics and by separating the DNA fragments by capillary electrophoresis.
4. Shotgun sequencing:
This type of sequencing was followed for sequencing genomes because in this method shotgun is utilized to form fragments from whole genome and then they are cloned and cultured. From the cloned sequence, sequencing carried out. After carrying out all the clones, sequence of the original genome was determined. In this method initially a long DNA sequence is broken down into a library of small fragments (0.5 -5.0 kb) produced by restriction enzymes or shear forces.
Then the small fragments are categorized according to their sizes and the appropriate cloning vectors are used to clone the fragments and produce multiple copies. The small cloned fragments are randomly selected and sequenced by dideoxy fluorescent sequencing technique using dye primers to label cloned fragments. Gel electrophoresis separates fragments by size. This step also removes the vector DNA from the cloned labeled fragments.
Labeled fragments are arranged by using PHred / PHrap analysis which is a computer program which determines the sequence of the fragments by examining the overlapped regions of the cloned DNA fragments to give the overall ds DNA sequence. The gap (no overlap) and single stranded portions in the DNA sequence is filled by either direct sequencing by using primer or using the best fit pieces from the library of cloned fragments.

4. Cycle Sequencing:
Cycle sequencing is a simple method in which successive rounds of denaturation, annealing and extension in a thermal cycler result in linear amplification of extension products. The products are then loaded onto a gel or injected into a capillary. All current ABI Prism DNA sequencing kits use cycle sequencing protocols.

Advantages of cycle sequencing:
RNA SEQUENCING:
RNA may be rapidly sequenced by only a slight modification of DNA sequencing procedures. The RNA to be sequenced is transcribed into a complementary strand of DNA (cDNA) through the action of RNA directed DNA polymerase also known as reverse transcriptase. The resulting cDNA may then be sequenced by either the chemical cleavage or the chain terminator method.
