NUCLEOTIDE SEQUENCE DATABASES (GENBANK, EMBL AND DDBJ)
Genbank:
GenBank is a comprehensive public database of nucleotide sequences and supporting bibliographic and biological annotation. GenBank is built and distributed by the National Center for Biotechnology Information (NCBI), a division of the National Library of Medicine (NLM), located on the campus of the US National Institutes of Health (NIH) in Bethesda, MD, USA. NCBI builds GenBank primarily from the submission of sequence data from authors and from the bulk submission of expressed sequence tag (EST), genome survey sequence (GSS) and other high-throughput data from sequencing centers. The US Office of Patents and Trademarks also contributes sequences from issued patents. GenBank participates with the European Molecular Biology Laboratory Nucleotide Sequence Database (EMBL) and the DNA Databank of Japan (DDBJ) as a partner in the International Nucleotide Sequence Database Collaboration (INSDC), which exchanges data daily to ensure that a uniform and comprehensive collection of sequence information is available worldwide.
Each GenBank entry includes a concise description of the sequence, the scientific name and taxonomy of the source organism, bibliographic references and a table of features (www.ncbi.nlm.nih.gov/collab/FT/index.html) listing areas of biological significance, such as coding regions and their protein translations, transcription units, repeat regions and sites of mutations or modifications. The files in the GenBank distribution have traditionally been partitioned into ‘divisions’ that roughly correspond to taxonomic groups, such as bacteria (BCT), viruses (VRL), primates (PRI) and rodents (ROD). In recent years, divisions have been added to support specific sequencing strategies. These include divisions for EST, GSS, high-throughput genomic (HTG), highthroughput cDNA (HTC) and environmental sample (ENV) sequences, making a total of 20 divisions. The newest division, Transcriptome Shotgun Assemblies (TSAs), was added in the past year and is described below. For convenience in file transfer, the GenBank data are partitioned into multiple files, currently more than 1600, for the bimonthly GenBank releases on the NCBI FTP site.
Each GenBank record, consisting of both a sequence and its annotations, is assigned a unique identifier called an accession number that is shared across the three collaborating databases (GenBank, DDBJ and EMBL). The accession number appears on the ACCESSION line of a GenBank record and remains constant over the lifetime of the record, even when there is a change to the sequence or annotation. Changes to the sequence data itself are tracked by an integer extension of the accession number, and this Accession.version identifier appears on the VERSION line of the GenBank flat file. The initial version of a sequence has the extension ‘.1’. In addition, each version of the DNA sequence is also assigned a unique NCBI identifier called a GI number that also appears on the VERSION line following the Accession.version:
ACCESSION AF000001
VERSION AF000001.1 GI: 987654321.
GenBank is accessible through the National Center for Biotechnology Information (NCBI) Entrez retrieval system, which integrates data from the major DNA and protein sequence databases along with taxonomy, genome, mapping, protein structure and domain information, and the biomedical journal literature via PubMed. Most submissions are made using the web-based BankIt or standalone Sequin programs, and accession numbers are assigned by GenBank_ staff upon receipt.
NCBI distributes GenBank releases in the traditional flat file format as well as in the ASN.1 format used for internal maintenance. The full bimonthly GenBank release along with the daily updates, which incorporate sequence data from EMBL and DDBJ, are available by anonymous FTP from NCBI at ftp.ncbi.nih.gov/genbank as well as from a mirror site at the University of Indiana (ftp://bio-mirror.net/biomirror/genbank/). To access GenBank and its related retrieval and analysis services, begin at the NCBI Homepage: www.ncbi.nlm.nih.gov.
EMBL (ENA):
Once made public, data files submitted to EMBL-Bank are available through the ENA Browser using REST URLs. Downloading of EMBL-Bank data is also supported through ftp. EMBL-Bank data is available for bulk download through FTP at ftp://ftp.ebi.ac.uk/pub/databases/embl/.
The European Nucleotide Archive (http://www.ebi.ac.uk/ena/) captures and presents information relating to experimental workflows that are based around nucleotide sequencing. A typical workflow includes the isolation and preparation of material for sequencing, a run of a sequencing machine in which sequencing data are produced and a subsequent bioinformatic analysis pipeline. ENA records this information in a data model that covers input information (sample, experimental setup, machine configuration), output machine data (sequence traces, reads and quality scores) and interpreted information (assembly, mapping, functional annotation).
Data arrive at ENA from a variety of sources. These include submissions of raw data, assembled sequences and annotation from small-scale sequencing efforts, data provision from the major European sequencing centres and routine and comprehensive exchange with our partners in the International Nucleotide Sequence Database Collaboration (INSDC).
Provision of nucleotide sequence data to ENA or its INSDC partners has become a central and mandatory step in the dissemination of research findings to the scientific community. ENA works with publishers of scientific literature and funding bodies to ensure compliance with these principles and to provide optimal submission systems and data access tools that work seamlessly with the published literature.
ENA is made up of a number of distinct databases that includes EMBL-Bank, the newly established Sequence Read Archive (SRA) and the Trace Archive each with their own data formats and standards.
Although the ENA has almost 30 years of history, the data and services are constantly changing to reflect growing volumes of data, ever improving sequencing technology and the broadening of applications to which sequencing is now put. As part of the global effort to improve access to and usability of nucleotide sequencing data, we collaborate extensively in the development of our services and technologies and in standards activities.
The ENA is developed and maintained at the EMBL-EBI under the guidance of the INSDC International Advisory Committee and a newly formed Scientific Advisory Board.
DDBJ:
DNA Data Bank of Japan (DDBJ) (http://www.ddbj.nig.ac.jp) collected and released 2 368 110 entries or 1 415 106 598 bases in the period from July 2007 to June 2008. Data can be obtained at the DDBJ ftp site (http://www.ddbj.nig.ac.jp/ftp_soap-e.html). It is located at the National Institute of Genetics (NIG) in the Shizuoka prefecture of Japan. DDBJ began data bank activities in 1986 at NIG and remains the only nucleotide sequence data bank in Asia. Although DDBJ mainly receives its data from Japanese researchers, it can accept data from contributors from any other country. DDBJ is primarily funded by the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT). DDBJ has an international advisory committee which consists of nine members, 3 members each from Europe, US, and Japan. This committee advises DDBJ about its maintenance, management and future plans once a year. Nucleotide sequence is in Genbank format.
In addition to traditional nucleotide data, the DDBJ has released raw sequencing data output from the DDBJ Trace Archive (DTA, http://trace.ddbj.nig.ac.jp/dta/dta_index_e.shtml) and the DDBJ Sequence Read Archive (DRA, http://trace.ddbj.nig.ac.jp). The DTA contains raw sequencing data obtained from gel/capillary platforms such as Applied Biosystems ABI 3730.
A secondary database is constructed by re-analyzing or modifying the primary data consisting of nucleotide sequence flat files released from the INSD. The DDBJ provides users with various types of secondary databases. DDBJ Amino Acid Database (DAD) records amino acid sequences extracted from values of /translation qualifiers in the nucleotide flat files. The DAD consists of 17348613 entries (4825871820 amino acids) as of June 2010. Gene Trek in Prokaryote Space (GTPS, http://gtps.ddbj.nig.ac.jp/) is a prokaryotic genome database that has been re-annotated by a sophisticated common protocol. GTPS assigns reliability grades to entire re-annotated protein-coding genes according to the result of blast and motif scans. GTPS can predict genes that are not annotated originally.
Next-generation sequencing platforms are gradually replacing the DNA microarray for measuring molecular abundances at the genomic scale. To accommodate quantitative genomics data from traditional and new platforms, the DDBJ has decided to launch a new archival database, the DDBJ Omics aRchive (DOR). The DOR has agreed to collaborate with ArrayExpress at the EBI to exchange data. The DOR archives data in compliance with two international guidelines, Minimum Information about a High-Throughput Sequencing Experiment (MINSEQE) and Minimum Information about a Microarray Experiment (MIAME), as ArrayExpress does. As NGSes are used to quantify DNA/RNA molecules, researchers submit their raw data to the DRA and their processed data to the DOR. The DOR will establish a submission brokering system in which researchers deposit necessary data sets to the DOR, and the raw data are automatically registered to the DRA.
PROTEIN SEQUENCE DATABASE (SWISS-PROT, TR-EMBL, PIR_PSD, EXPASY)
UniProt is a comprehensive, high-quality and freely accessible database of protein sequence and functional information, many entries being derived from genome sequencing projects. It contains a large amount of information about the biological function of proteins derived from the research literature.
The UniProt consortium comprises the European Bioinformatics Institute (EBI), the Swiss Institute of Bioinformatics (SIB), and the Protein Information Resource (PIR). EBI, located at the Wellcome Trust Genome Campus in Hinxton, UK, hosts a large resource of bioinformatics databases and services. SIB, located in Geneva, Switzerland, maintains the ExPASy (Expert Protein Analysis System) servers that are a central resource for proteomics tools and databases. PIR, hosted by the National Biomedical Research Foundation (NBRF) at the Georgetown University Medical Center in Washington, DC, USA, is heir to the oldest protein sequence database, Margaret Dayhoff's Atlas of Protein Sequence and Structure, first published in 1965. In 2002, EBI, SIB, and PIR joined forces as the UniProt consortium.
Each consortium member is heavily involved in protein database maintenance and annotation. Until recently, EBI and SIB together produced the Swiss-Prot and TrEMBL databases, while PIR produced the Protein Sequence Database (PIR-PSD). These databases coexisted with differing protein sequence coverage and annotation priorities.
Swiss-Prot was created in 1986 by Amos Bairoch during his PhD and developed by the Swiss Institute of Bioinformatics and subsequently developed by Rolf Apweiler at the European Bioinformatics Institute. Swiss-Prot aimed to provide reliable protein sequences associated with a high level of annotation (such as the description of the function of a protein, its domain structure, post-translational modifications, variants, etc.), a minimal level of redundancy and high level of integration with other databases. Recognizing that sequence data were being generated at a pace exceeding Swiss-Prot's ability to keep up, TrEMBL (Translated EMBL Nucleotide Sequence Data Library) was created to provide automated annotations for those proteins not in Swiss-Prot. Meanwhile, PIR maintained the PIR-PSD and related databases, including iProClass, a database of protein sequences and curated families. The consortium members pooled their overlapping resources and expertise, and launched UniProt in December 2003
Uniprot:
UniProt provides four core databases: UniProtKB (with sub-parts Swiss-Prot and TrEMBL), UniParc, UniRef, and UniMes.
UniProt Knowledgebase (UniProtKB) is a protein database partially curated by experts, consisting of two sections: UniProtKB/Swiss-Prot (containing reviewed, manually annotated entries) and UniProtKB/TrEMBL (containing unreviewed, automatically annotated entries). As of 3 October 2012, release "2012_09" of UniProtKB/Swiss-Prot contains 538,010 sequence entries (comprising 190,998,508 amino acids abstracted from 213,490 references) and release "2012_09" of UniProtKB/TrEMBL contains 26,079,526 sequence entries (comprising 8,448,404,066 amino acids).
Swiss-Prot
Swiss-Prot is a high-quality, manually annotated, non-redundant protein sequence database. It combines information extracted from scientific literature and biocurator-evaluated computational analysis. The aim of UniProtKB/Swiss-Prot is to provide all known relevant information about a particular protein. Annotation is regularly reviewed to keep up with current scientific findings. The manual annotation of an entry involves detailed analysis of the protein sequence and of the scientific literature.
Sequences from the same gene and the same species are merged into the same database entry. Differences between sequences are identified, and their cause documented (for example alternative splicing, natural variation, incorrect initiation sites, incorrect exon boundaries, frameshifts, unidentified conflicts). A range of sequence analysis tools is used in the annotation of UniProtKB/Swiss-Prot entries. Computer-predictions are manually evaluated, and relevant results selected for inclusion in the entry. These predictions include post-translational modifications, transmembrane domains and topology, signal peptides, domain identification, and protein family classification. Relevant publications are identified by searching databases such as PubMed. The full text of each paper is read, and information is extracted and added to the entry. Annotation arising from the scientific literature includes, but is not limited to:
Annotated entries undergo quality assurance before inclusion into UniProtKB/Swiss-Prot. When new data becomes available, entries are updated.
TrEMBL
TrEMBL contains high-quality computationally analyzed records, which are enriched with automatic annotation. It was introduced in response to increased dataflow resulting from genome projects, as the time- and labour-consuming manual annotation process of UniProtKB/Swiss-Prot could not be broadened to include all available protein sequences. The translations of annotated coding sequences in the EMBL-Bank/GenBank/DDBJ nucleotide sequence database are automatically processed and entered in UniProtKB/TrEMBL. UniProtKB/TrEMBL also contains sequences from PDB, and from gene prediction, including Ensembl, RefSeq and CCDS.
UniParc
UniProt Archive (UniParc) is a comprehensive and non-redundant database, which contains all the protein sequences from the main, publicly available protein sequence databases. Proteins may exist in several different source databases, and in multiple copies in the same database. In order to avoid redundancy, UniParc stores each unique sequence only once. Identical sequences are merged, regardless of whether they are from the same or different species. Each sequence is given a stable and unique identifier (UPI), making it possible to identify the same protein from different source databases. UniParc contains only protein sequences, with no annotation. Database cross-references in UniParc entries allow further information about the protein to be retrieved from the source databases. When sequences in the source databases change, these changes are tracked by UniParc and history of all changes is archived.
UniRef
The UniProt Reference Clusters (UniRef) consist of three databases of clustered sets of protein sequences from UniProtKB and selected UniParc records. The UniRef100 database combines identical sequences and sequence fragments (from any organism) into a single UniRef entry. The sequence of a representative protein, the accession numbers of all the merged entries and links to the corresponding UniProtKB and UniParc records are displayed. UniRef100 sequences are clustered using the CD-HIT algorithm to build UniRef90 and UniRef50. Each cluster is composed of sequences that have at least 90% or 50% sequence identity, respectively, to the longest sequence. Clustering sequences significantly reduces database size, enabling faster sequence searches.
UniMES
The UniProt Metagenomic and Environmental Sequences (UniMES) database is a repository specifically developed for metagenomic and environmental data. The predicted proteins from this dataset are combined with automatic classification by InterPro to enhance the original information with further analysis.
UniProtKB contains protein sequences from known species, data arising from metagenomics studies is from environmental (i.e., uncultured) samples and as such the species may not be known or as yet identified. UniMES was developed for this data. Data from UniMES is not included in UniProtKB or UniRef, but is included in UniParc. As of July 2012, UniMES contains only data from the Global Ocean Sampling Expedition (GOS). The environmental sample data contained within this database is not present in either the UniProt Knowledgebase or the UniProt Reference Clusters.
Download site: ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/
ftp://ftp.uniprot.org/pub/databases/uniprot/
PIR:
The Protein Information Resource (PIR), located at Georgetown University Medical Center (GUMC), is an integrated public bioinformatics resource to support genomic and proteomic research, and scientific studies.
The Protein Information Resource (PIR) is an integrated public resource of protein informatics that supports genomic and proteomic research and scientific discovery. PIR maintains the Protein Sequence Database (PSD), an annotated protein database containing over 283 000 sequences covering the entire taxonomic range. Family classification is used for sensitive identification, consistent annotation, and detection of annotation errors. The superfamily curation defines signature domain architecture and categorizes memberships to improve automated classification. To increase the amount of experimental annotation, the PIR has developed a bibliography system for literature searching, mapping, and user submission, and has conducted retrospective attribution of citations for experimental features. PIR also maintains NREF, a non-redundant reference database, and iProClass, an integrated database of protein family, function, and structure information. PIR-NREF provides a timely and comprehensive collection of protein sequences, currently consisting of more than 1 000 000 entries from PIR-PSD, SWISS-PROT, TrEMBL, RefSeq, GenPept, and PDB. The PIR web site (http://pir.georgetown.edu) connects data analysis tools to underlying databases for information retrieval and knowledge discovery, with functionalities for interactive queries, combinations of sequence and text searches, and sorting and visual exploration of search results. The FTP site provides free download for PSD and NREF biweekly releases and auxiliary databases and files.
PIR-PSD:
The website address is http://pir.georgetown.edu/pirwww/dbinfo/pir_psd.shtml
Superfamily classification and curation. A unique characteristic of the PIR-PSD is the superfamily classification that provides comprehensive, non-overlapping, and hierarchical clustering of sequences to reflect their evolutionary relationships. To further improve the quality of automated classification, we have conducted systematic superfamily curation that: (i) defines the signature domain architecture (number, order, and types of domains) characteristic of the superfamily, (ii) categorizes regular and associate members to distinguish sequence entries sharing the signature features from outliers (such as fragments), and (iii) designates representative and seed members amongst regular members. Several thousand superfamilies have been manually curated. The seed members provide a basis for automatic placement of new sequences into existing superfamilies and for automatic generation of multiple sequence alignments and phylogenetic trees. Currently, over 99% of PSD sequences are classified into families of closely related sequences (at least 45% identical), and over two-thirds of sequences are classified into >36 000 superfamilies. Bibliography mapping and attribution. To improve the quality of protein annotation by increasing the amount of experimentally verified data with source attribution, the PIR has developed a bibliography information system and conducted retrospective attribution of literature data. The bibliography system allows browsing and searching of extensive literature collected for all protein entries from PubMed and other curated molecular databases, together with an interface for scientists to categorize and submit literature information for mapped proteins.
In PIR-PSD, protein features such as binding sites, structural motifs, and post-translational modifications are tagged with ‘experimental’ status for experimentally determined features to distinguish from those that are computationally predicted; however, they had not been associated with literature citations. A systematic manual attribution of experimental features is being carried out with computer-assisted mapping to existing protein bibliographic information. So far, a few thousand experimental features have been associated with publications.
Both PSD and NREF XML distributions have an associated DTD (Document Type Definition) file. The sequence files of both databases are distributed in FASTA format.
The downloading site was ftp://ftp.pir.georgetown.edu/pir_databases/.
PIR tools:
The PIR web site connects data mining and sequence analysis tools to underlying databases for information retrieval and knowledge discovery, with functionalities for interactive queries, combinations of sequence and annotation text searches, and sorting and visual exploration of search results. The three major databases (PSD, NREF and iProClass) represent primary entry points in the PIR web site, all of which provide text search for entry and list retrieval as well as BLAST search and peptide match. Direct entry report retrieval is based on sequence unique identifiers of all underlying databases, such as PIR, SWISS-PROT, or RefSeq. Basic and advanced text searches return protein entries listed in summary lines with information on protein IDs, matched fields, protein name, taxonomy, superfamily, domain, and motif, with hypertext links to the full entry report and to cross-referenced databases. More than 50 fields are searchable, including about 30 database unique identifiers (e.g., PDB ID, EC number, PubMed ID, and KEGG pathway number) and a wide range of annotation texts (e.g., protein name, organism name, sequence feature, and paper title). The BLAST search and peptide search likewise return lists of matched entries with summary lines that also contain search statistics and matched sequence region. Protein entries returned from text and sequence searches can be selected for further analysis, including BLAST and FASTA search, pattern match, hidden Markov model (HMMER) domain search, ClustalW multiple sequence alignments and Phylip phylogentic tree generation, and graphical display of superfamily, domain and motif relationships. Species-based browsing and searching are supported for about 100 organisms, including over 70 complete genomes. The related sequences in FASTA clusters are retrievable based on sequence unique identifiers where neighbors are listed with annotation information and graphical display of matched sequence region.
Release 80.00 (31 Dec 2004) is the final release for the PIR-International Protein Sequence Database (PIR-PSD), the world's first database of classified and functionally annotated protein sequences that grew out of the Atlas of Protein Sequence and Structure (1965-1978) edited by Margaret Dayhoff. Produced and distributed by the Protein Information Resource in collaboration with MIPS (Munich Information Center for Protein Sequences) and JIPID (Japan International Protein Information Database), PIR-PSD has been the most comprehensive and expertly-curated protein sequence database in the public domain for over 20 years. In 2002, PIR joined EBI (European Bioinformatics Institute) and SIB (Swiss Institute of Bioinformatics) to form the UniProt consortium. PIR-PSD sequences and annotations have been integrated into UniProt Knowledgebase. Bi-directional cross-references between UniProt (UniProt Knowledgebase and/or UniParc) and PIR-PSD are established to allow easy tracking of former PIR-PSD entries. PIR-PSD unique sequences, reference citations, and experimentally-verified data can now be found in the relevant UniProt records.
For any help contact pirmail@georgetown.edu for information regarding PIR. PIR has recently joined forces with the European Bioinformatics Institute and the Swiss Institute of Bioinformatics to establish the Universal Protein Resource (UniProt), the central resource of protein sequence and function. Please submit your sequences directly to UniProtKB using SPIN, the new web-based tool for submitting directly sequenced proteins. SPIN is the web-based tool for submitting directly sequenced protein sequences and their biological annotations to the UniProt Knowledgebase. SPIN guides you through a sequence of forms allowing interactive submission. The information required to create a database entry will be collected during this process. The website address is http://www.ebi.ac.uk/swissprot/Submissions/spin/index.jsp.
EXPASY:
The ExPASy (the Expert Protein Analysis System) World Wide Web server (http://www.expasy.org), is provided as a service to the life science community by a multidisciplinary team at the Swiss Institute of Bioinformatics (SIB). It provides access to a variety of databases and analytical tools dedicated to proteins and proteomics. ExPASy databases include SWISSPROT and TrEMBL, SWISS-2DPAGE, PROSITE, ENZYME and the SWISS-MODEL repository. Analysis tools are available for specific tasks relevant to proteomics, similarity searches, pattern and profile searches, post-translational modification prediction, topology prediction, primary, secondary and tertiary structure analysis and sequence alignment. These databases and tools are tightly interlinked: a special emphasis is placed on integration of database entries with related resources developed at the SIB an elsewhere, and the proteomics tools have been designed to read the annotations in SWISS-PROT in order to enhance their predictions. ExPASy started to operate in 1993, as the first WWW server in the field of life sciences. In addition to the main site in Switzerland, seven mirror sites in different continents currently serve the user community.
Databases:
ExPASy is the main host for the following databases that are partially or completely developed at the SIB in Geneva:
The SWISS-PROT knowledgebase (http://www.expasy.org/sprot/) is a curated protein sequence database, which strives to provide high quality annotations (such as the description of the function of a protein, its domain structure, post-translational modifications and variants), a minimal level of redundancy and a high level of integration with other databases. SWISS-PROT is supplemented by
TrEMBL, which contains computer-annotated entries for all sequences not yet integrated in SWISS-PROT. SWISS-PROT and TrEMBL are maintained collaboratively by the SIB and the European Bioinformatics Institute (EBI).
SWISS-2DPAGE (http://www.expasy.org/ch2d/) is a database of proteins identified on two-dimensional polyacrylamide gel electrophoresis (2D PAGE). SWISS-2DPAGE contains data from a variety of human and mouse biological samples as well as from Arabidopsis thaliana, Escherichia coli, Saccharomyces cerevisiae and Dictyostelium discoideum.
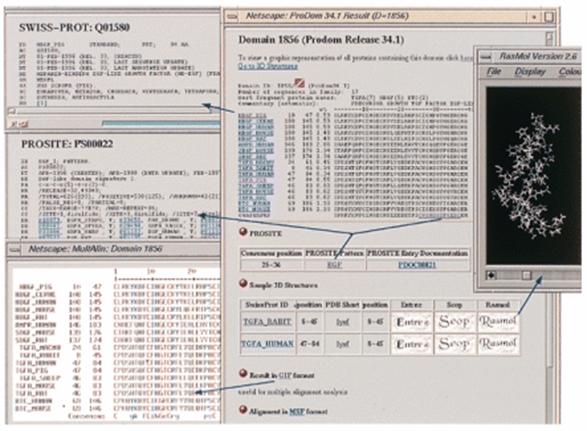
PROSITE (http://www.expasy.org/prosite/) is a database of protein domains and families. PROSITE contains biologically significant sites, patterns and profiles that help to reliably identify to which known protein family a new sequence belongs.
ENZYME (http://www.expasy.org/enzyme/) is a repository of information relative to the nomenclature of enzymes.

SWISS-MODEL Repository (http://www.expasy.org/swissmod/smrep.html) is a database of automatically generated structural protein models.
Recently Uniprot also included in ExPASy.
SWISS-PROT, PROSITE, ENZYME and SWISS-2DPAGE are updated at a frequency of _1–2 weeks. For all the ExPASy databases, data and associated documentation files can be copied locally by anonymous FTP (ftp.expasy.org). In particular, the different download options for the SWISS-PROT and TrEMBL databases, including the different available subsections, release frequencies and data formats, are documented at http://www.expasy.org/sprot/download.html.
A large variety of documents (user manual, release notes, indices, nomenclature documents, etc.) are available with SWISS-PROT; these documents can all be browsed from ExPASy (http://www.expasy.org/sprot/sp-docu.html) and are enhanced by a variety of hyperlinks.
Online tools:
Online tools are either targeted toward the access and display of the databases or can be used to analyze protein sequences and proteomics data originating from 2D-PAGE and mass spectrometry experiments. These tools can all be accessed from ExPASy (http://www.expasy.org/tools/).
Sequence analysis tools
BLAST provides very fast similarity searches of a protein sequence against a protein or nucleotide database. The ExPASy BLAST service is maintained in collaboration with the Swiss EMBnet node on dedicated hardware. The native output of BLAST is extended with several original features.
ScanProsite scans a sequence against all the patterns, profiles and rules in PROSITE or scans a pattern, profile or rule against all sequences in SWISS-PROT, TrEMBL and/or PDB.
SWISS-MODEL is an automated knowledge-based protein modelling server. It is able to build models for the 3D structure of proteins whose sequence is closely related to that of proteins with known 3D structure.
ProtParam calculates physico-chemical parameters of a protein sequence such as the amino acid composition, the pl, the atomic composition, the extinction coefficient, etc.
ProtScale computes and represents the profile produced by any amino acid scale on a selected protein. Some 50 predefined scales are available, such as the Doolittle and Kyte hydrophobicity scale.
RandSeq generates a random protein sequence, based on a user-specified amino acid composition and sequence length.
Sulfinator predicts tyrosine sulfation sites within protein sequences.
Translate translates a nucleotide sequence into a protein in six reading frames.
Proteomics tools
AACompIdent identifies a protein by its amino acid composition.
AACompSim finds for a given SWISS-PROT entry, the database entries which have the most similar amino acid composition.
Compute pI/MW computes the theoretical isoelectric point (pI) and molecular weight (MW) from a SWISS-PROT or TrEMBL entry or for a user sequence.
FindMod predicts potential protein post-translational modifications and potential single amino acid substitutions in peptides. Experimentally measured peptide masses are compared with the theoretical peptides calculated from a specified SWISS-PROT entry or from a user-entered sequence. Mass differences are used to better characterize the protein of interest.
FindPept identifies peptides resulting from unspecific cleavage of proteins by their experimental masses, taking into account artefactual chemical modifications, posttranslational modifications and protease autolytic cleavage.
GlycanMass calculates the mass of an oligosaccharide structure.
GlycoMod predicts possible oligosaccharide structures that occur on proteins from their experimentally determined masses. This is done by comparing the mass of a potential glycan to a list of pre-computed masses of glycan compositions.
PeptideCutter predicts potential protease cleavage sites and sites cleaved by chemicals in a given protein sequence.
PeptideMass calculates the theoretical masses of peptides generated by the chemical or enzymatic cleavage of proteins so as to assist in the interpretation of peptide mass fingerprinting.
PeptIdent, TagIdent, MultiIdent, these three related programs identify proteins using a variety of experimental information such as the pI, theMW, the amino acid composition, partial sequence tags and peptide mass fingerprinting data.
Softwares:
DeepView (SWISS-PdbViewer) (spdbv) (http://www.expasy.org/spdbv/) is an application running on the Microsoft Windows, Mac, SGI and Linux platforms, offering a wide range of options to visualize and manipulate protein structures. It can also be used as a WWW helper application for the display of PDB formatted entries. Swiss-PdbViewer can be downloaded from ExPASy and complements the aforementioned SWISS-MODEL homology-modeling tool.
LALNVIEW (http://www.expasy.org/tools/lalnview.html) is an application that runs on the Microsoft Windows, Mac and Unix platforms. LALNVIEW is a graphical viewer for pairwise sequence alignments. It can be used to display the results of a pairwise alignment carried out with the SIM software also installed on ExPASy (http://www.expasy.org/tools/sim-prot.html).
Any questions and feedback on ExPASy should be reported using the ExPASy helpdesk on the Support page (http://www.expasy.org/support).
DERIVED DATABASES (PROSITE, PRODOM, PFAM, PRINTS)
PROSITE:
The PROSITE database uses two kinds of signatures or descriptors to identify conserved regions, i.e. patterns and generalized profiles, which both have their own strengths and weaknesses defining their area of optimum application. Each PROSITE signature is linked to an annotation document where the user can find information on the protein family or domain detected by the signature: origin of its name, taxonomic occurrence, domain architecture, function, 3D structure, main characteristics of the sequence, domain size and some references.
The documentation page has also been reorganized. It nowcontains three main sections:
(i) The description part that exposes the main characteristics of the domain or the family and a representative list of proteins that contain the domain or belong to the family. 105
(ii) A technical section that refers to the descriptors used to identify the domain or family. For each descriptor, there is a link to a domain architecture view of UniProtKBproteins
matched by the descriptor, an MSA in different formats, a link to retrieve the list of proteins matched by the descriptor in various formats and a link to a taxonomy tree view of all entries containing the domain. There is also an external link to MSDsite to view ligand binding statistics of the domain and a link to 3D structures.
(iii) The third section is the reference block where, for each reference, we added the PubMed ID and a direct link to the article.

The PROSITE database is now complemented by a series 15 of rules that can give more precise information about specific residues. During the last 2 years, the documentation and the ScanProsite web pages were redesigned to add more functionality. The latest version of PROSITE (release 19.11 of September 27, 2005) contains 1329 patterns and 552 profile entries. Over the past 2 years more than 200 domains have been added, and now 52% of UniProtKB/Swiss-Prot entries (release 48.1 of September 27, 2005) have a cross-reference to a PROSITE entry. The database is accessible at http://www.expasy.org /prosite/.
Patterns or regular expressions are useful tools to identify short and well-conserved regions, such as catalytic sites, binding sites, post-transcriptional modifications (PTMs) or zinc fingers. We have developed a tool to identify weak patterns and automatically update them. This tool uses the PROSITE match list, which stores true positives, false positives (FP), false negatives (FN), partial and unknown matches, to generate a new pattern that minimizes FP and FN.
FP and FN updates are treated independently. We first take care of FN in a three-step procedure:
(i) The patterns that can potentially be updated are selected. Updating a pattern to recover FN amounts to introduce more variability in the pattern, but it increases the risk of creating new FP. Hence, only patterns that are stringent enough can be updated. The selection procedure consists of running all PROSITE patterns on a random database to keep only the ones that do not produce too many matches.
(ii) Mismatches produced by each FN are detected and the pattern is modified accordingly to accept the observed residues.
(iii) The new pattern is tested on a random database to see whether it is still stringent enough. If it produces too many matches in a random database, the pattern is refined and some mismatch positions are removed.
To remove false positives we check ‘wildcard’ positions (‘x’ with the PROSITE syntax) in the pattern. We look at these positions for amino acids that are only found in FP sequences. These amino acids are then ‘forbidden’ ({} with the PROSITE syntax) at these positions in the new pattern. The new pattern is then used to scan Swiss-Prot and all new matches are checked manually. Only patterns that produce no new false positives are kept. This strategy has allowed the automatic update of 943 patterns (out of a total of 1322 patterns in PROSITE). 2661 FN (out of a total of 14 412) and 1927 FP (out of a total of 7446) were removed. The application of these two strategies allowed a decrease of the number of FP and FN in the Swiss-Prot part of UniProt by 25%.
There are currently several tools to construct efficient profiles based on MSA (8). All these tools were designed to recover very divergent proteins (<20% of similarity). They were developed 10 years ago when protein databases were quite small and very few representative genomes were sequenced.
Profile builder parameters can then be adjusted according to the annotation. We have used this strategy to adjust specific parameters in a column-dependant manner. We have tested the weight of the matrix, gap and insertion penalties. The tool aim is to be more stringent on specific columns and to produce a better local alignment, which then helps to re-localize the functional residues in sequences matched by the profile.
The PROSITE website was redesigned and new predictive tools were implemented to assign more detailed functional information to the scanned proteins. Users who want to scan their own proteins against all PROSITE entries or to scan a PROSITE entry against a protein database will find a new version of the ScanProsite web page.
(i) A pattern or regular expression is a quantitative descriptor: it either matches or does not. Therefore a good pattern is usually located in a short well-conserved region. Such regions are typically enzyme catalytic sites, prosthetic group attachment sites (haem, pyridoxal phosphate, biotin, etc.), metal ion binding amino acids, cysteines involved in disul®de bonds or regions involved in binding a molecule. Even though the scope of a regular expression is limited to these particular biological regions, patterns are still very popular because of their intelligibility for users.
(ii) A pro®le is a table of position-speci®c amino acid weights and gap costs. Various methods can be used to ®ll a pro®le table from a multiple alignment. Most frequently, a substitution matrix is used to convert a residue frequency distribution into weights, but alternative methods can be applied including structure-based approaches and methods involving hidden Markov modelling. These weights (also referred to as scores) are used to calculate a similarity score for any alignment between a pro®le and a sequence, or part of a pro®le and a sequence. An alignment with a similarity score higher than or equal to a given threshold value constitutes a motif occurrence. This threshold is estimated by calibrating the pro®le against a randomized protein database. The normalization procedure used for PROSITE pro®les makes the normalized scores independent of the database size, allowing the comparison of scores from different searches. The quantitative behaviour of a pro®le allows the acceptance of a mismatch at a highly conserved position if the rest of the sequence displays a suf®ciently high level of similarity and therefore allows the detection of poorly conserved domains such as immunoglobulin, SH2 or SH3. Another advantage of pro®les over patterns is that they are not con®ned to small regions with high sequence similarity. Rather, they attempt to characterize a protein family or domain over its entire length.
ftp://ftp.expasy.org/databases/prosite/release_with_ updates/
info@genebio.com
Geneva Bioinformatics
(GeneBio) S.A, Case Postale 210, CH-1211 Geneva, 12, Switzerland
PRODOM:
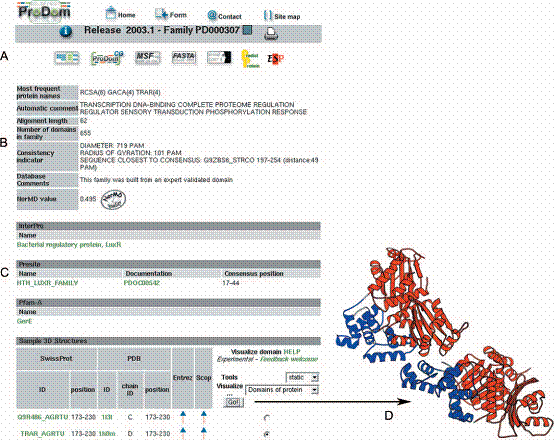
ProDom is a comprehensive database of protein domain families generated from the global comparison of all available protein sequences. The ProDom database contains protein domain families generated from the SWISS-PROT database by automated sequence comparisons. Recent improvements include the use of three-dimensional (3D) information from the SCOP database; a completely redesigned web interface (http://www.toulouse.inra.fr/prodom.html); visualization of ProDom domains on 3D structures; coupling of ProDom analysis with the Geno3D homology modeling server; Bayesian inference of evolutionary scenarios for ProDom families. In addition, we have developed ProDom-SG, a ProDom-based server dedicated to the selection of candidate proteins for structural genomics.
ProDom ‘domains’ thus essentially reflect protein subsequences conserved in various proteins. For each domain family a multiple alignment and a consensus sequence are computed, as well as links to PROSITE and PDB where relevant. We have set up a World Wide Web server (http://protein.toulouse.inra.fr/prodom.html. ) which provides graphical access to ProDom. It allows the user to get a schematic visualisation of all proteins sharing a given homologous domain, or all proteins sharing a homologous domain with a given protein. Hypertext links give access to multiple alignments, consensus sequences and PROSITE and PDB links for each domain family. Any query sequence can be compared against ProDom using the BLAST or the WU-BLAST algorithm with a graphical output: a possible decomposition of new protein sequences into domains is quickly visualised.
ProDom is built as two text files, ‘prodom.mul’ and ‘prodom’. Each entry is a domain family with an automatically generated comment and a multiple domain alignment in the ‘prodom.mul’ file, or a consensus sequence in the ‘prodom’ file. We also provide a tool (FETCHDOM) to retrieve the domain decomposition of any protein that is present in ProDom, or to fetch multiple alignments of ProDom domain families.
The main ProDom form consists of two parts. The first part (ProDom Browsing) allows
querying of ProDom in a variety of ways: (i) by accession number (Display a ProDom entry); (ii) by the display of all proteins belonging to one or several ProDom families with logical AND/OR operators (All proteins in ProDom families); (iii) by related databases (InterPro, PROSITE, PFAM or PDB); (iv) by SWISS-PROT/TrEMBL identifier or accession number; and (v) by keyword search with AND/OR operators. The output is either information on a given domain family (Figure 1) or cartoons displaying the domain arrangements of all proteins matching the query (Figure 2). The number of different cartoons available for domain display was increased from 14 160 to 237 888 with the use of 64 colours, providing for more legible outputs while preserving consistency across different displays. The second part of the main ProDom form allows for BLAST searches in ProDom (Compare your sequence with ProDom), suggesting a possible domain arrangement for any query protein. When 3D structures are available for target domains, the output is directly linked to both SWISSMODEL and Geno3D servers for homology-based domain modelling.
ProDom-CG is a subset of ProDom, restricted to sequences derived from completely sequenced genomes. Bacterial protein sets were retrieved from the ExPASy server (ftp://www.expasy.org/databases/hamap/complete_proteomes), while eukaryotic protein sets were retrieved from the EBI server (http://www.ebi.ac.uk/integr8). All relevant multiple alignments and characteristics were recalculated on the resulting families. The taxonomy tree encompassing completely sequenced genomes was colour-coded so as to indicate ancestral nodes predicted to contain domains in a given ProDom-CG family. These colour-coded trees are available for each ProDom-CG entry on the ProDom website.
ProDom-SG FOR STRUCTURAL GENOMICS
ProDom-SG (Structural Genomics) server, designed to assist in the selection of protein domain families corresponding to potentially new folds on the basis of lack of detected homology. The server also allows for the identification of favourable protein candidates for crystallization studies. ProDom-SG was built in three steps. In the first step, only ProDom families with norMD values above 0.5 were considered. In the second step, potential homology relationships between ProDom families were identified using PSI-BLAST with family specific, position-specific scoring matrices. When applicable, the existence of such related families is indicated using a specific logo appearing at the top of the family information sheet. In the third step, both direct and indirect links to the PDB were recorded for each family.
Molscript is a program for displaying molecular 3D structures, such as proteins, in both schematic and detailed representations.
ProDom can be searched for similarity with a query sequence using BLAST tools (BLASTP, BLASTX or WU-BLAST). If the query sequence shares homology with at least one ProDom family, BLAST results are followed by a graphical representation of its proposed domain arrangement. Each target ProDom domain can be further exploited, either to align the query with the ProDom domain family using MultAlin, or to generate 3-D models of domains on the basis of homology using the Swiss-Model server, where applicable. A graphical view presents domain arrangements for a given protein, for proteins sharing a given ProDom domain, or for all proteins sharing homology with a given SWISS-PROT entry.
ftp://ftp.toulouse.inra.fr/pub/prodom
prodom@toulouse.inra.fr.
PFAM:
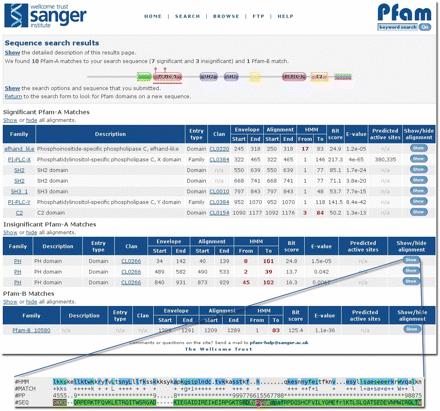
The Pfam database is a large collection of protein families, each represented by multiple sequence alignments and hidden Markov models (HMMs). The website is http://pfam.sanger.ac.uk/.
The Pfam database is one the most important collections of information in the world for classifying proteins. The database categorises 75 per cent of known proteins to form a library of protein families - a 'periodic table' of biology. The open access resource was established at the Wellcome Trust Sanger Institute in 1998. Its vision is to provide a tool which allows experimental, computational and evolutionary biologists to classify protein sequences and answer questions about what they do and how they have evolved. The Pfam project is led by Dr Alex Bateman at the Sanger Institute.
Pfam is a comprehensive database of conserved protein families. This collection of nearly 12 000 families is used extensively throughout the biological sciences, by experimental biologists researching specific proteins, computational biologists who need to organise sequences, and evolutionary biologists considering the origin and evolution of proteins. Pfam is also widely used in the structural biology community for identifying interesting new targets for structure determination.
From its inception 12 years ago, Pfam has been designed to scale with the growth in the number of new protein sequences deposited. Scalability is achieved by having a set of seed alignments, with each alignment containing a representative set of sequences that are relatively stable between releases of the database. The seed alignments are used to build profile hidden Markov models (HMMs) that can be used to search any sequence database for homologues in a sensitive and accurate fashion. Those homologues that score above the curated inclusion thresholds are aligned against the profile to make a full alignment.
Our goal is to make Pfam a comprehensive and accurate classification of all known protein sequences. The 11 912 curated families are known as Pfam-A and are found in approximately three quarters of known proteins. In order to increase our coverage further, we augment the Pfam-A family collection with a set of automatically generated families called Pfam-B. Pfam-B is derived from the ADDA domain collection, which is described later.
Pfam 24.0 represents a 24% increase in the total number of families, relative to Pfam 23.0. Most of these new families have come from one of two sources: (i) a family seeded by a structure deposited in the Protein Data Bank—wwPDB—that Pfam 23.0 did not cover, and (ii) Pfam-B families that we have used as a starting point for building Pfam-A, focusing particularly on Pfam-B clusters without a corresponding annotated family in InterPro. In addition to these, many families have been contributed via suggestions from the community.
ADDA is a method for automatically predicting protein sequence domains from protein sequence alignments alone. Briefly, the ADDA algorithm takes a set of non-redundant sequences and aligns them all-versus-all using BLAST. Sequences are then partitioned into domains by optimising an objective function that penalises domains that (i) split alignments or (ii) overlap with alignments only partially. The resultant domains are grouped into clusters using pairwise profile–profile comparisons. The whole procedure is calibrated using SCOP domains as a gold-standard.
Ten years ago, a family with more than 1000 sequences was considered to be large. Today, a growing number of families contain over 100 000 sequences. Depositions from large-scale metagenomic and other sequencing projects mean that we can expect the number of known sequences to grow into the billions, from the millions that we currently have. In order to deal with this explosion in the number of known sequences, we have made fundamental changes to the Pfam infrastructure. The most important of these has been the move to a new version of the profile HMM software, HMMER (http://hmmer.janelia.org/), which we use to build and search our models. Since 1998, Pfam (version 3.0 onwards) has utilised the HMMER2 package for building profile HMMs and searching them against sequences in the underlying sequence database. The new version of HMMER (version 3) is 100 times faster than the previous version and shows increased sensitivity.
The HMMER3 project has four main aims: (i) to adopt log-odds likelihood scores summed over alignment uncertainty (Forward scores) in place of optimal alignment (Viterbi) scores; (ii) to report posterior probabilities of alignment confidence; (iii) to be able to accurately and quickly calculate expectation values (E-values) for Forward scores (a previously unsolved problem); and (iv) to accelerate previous profile HMM performance by two orders of magnitude and achieve an overall speed competitive with BLAST.
ftp://ftp.sanger.ac.uk/pub/databases/Pfam/
rdf@sanger.ac.uk
PRINTS:
The PRINTS database houses a collection of protein fingerprints. These may be used to assign uncharacterized sequences to known families and hence to infer tentative functions. The September 2002 release (version 36.0) includes 1800 fingerprints, encoding _11 000 motifs, covering a range of globular and membrane proteins, modular polypeptides and so on. By contrast with PROSITE, which uses single consensus expressions to characterise particular families, PRINTS exploits groups of motifs to build characteristic signatures. These signatures offer improved diagnostic reliability by virtue of the mutual context provided by motif neighbours.
Fingerprints are groups of conserved sequence motifs that together provide diagnostic signatures for protein families. They derive much of their potency from the context afforded by multiple-motif matching, making them more flexible and powerful than single-motif approaches. Unlike some other pattern-matching methods, fingerprinting is well-suited to the creation of ‘hierarchical’ discriminators—e.g. this approach has been used to resolve G protein-coupled receptor (GPCR) super-families into their constituent families and receptor subtypes, and to sub-classify a variety of channel proteins, transporters and enzymes.

PRINTS was originally built as a single ASCII (text) file. To facilitate maintenance, we later developed a relational version of the resource, known as PRINTS-S.
PRINTS is released in major and minor versions: minor releases reflect updates, bringing the contents in line with the current version of the source database [a SWISS-PROT/
TrEMBL composite]; major releases denote the addition of new material to the resource. The latter are made quarterly, each release including 50 new annotated families. Four major releases have been made since the last report.
The tools available for searching PRINTS are: (i) a BLAST server, for searches against sequences matched in the current version of the database; and (ii) the FingerPRINTScan suite, for searches against fingerprints.
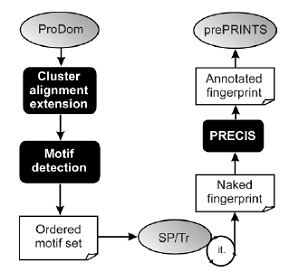
The growth of PRINTS is limited by the fact that it is maintained entirely manually, and hence it lags behind databases that are produced automatically. To begin to address this problem, we migrated the resource to a relational database management system. Although this facilitates routine maintenance and reduces some of the manual burdens, it does little to address database growth. We, therefore, developed an automatic supplement to PRINTS, termed prePRINTS (http://www.bioinf.man.ac.uk/prePRINTS/).
This exploits an automatic pipeline, which uses as input protein family clusters from ProDom. Motifs are detected automatically using a suite of programs, including DIALIGN and CLUSTALW, and are used to search a SWISS-PROT/TrEMBL composite database in an iterative fashion. Naked fingerprints generated by this process are then annotated automatically using PRECIS [Protein Reports Engineered from Concise Information in SWISS-PROT http://www.bioinf.man.ac.uk/cgi-bin/dbbrowser/precis/precis.cgi]. Finally, annotated fingerprints are deposited into a relational database.
http://www.bioinf.man.ac.uk/dbbrowser/PRINTS/index.php
ftp://ftp.bioinf.man.ac.uk/pub/prints
teresa.k.attwood@manchester.ac.uk
SEQUENCE SUBMISSION METHODS AND TOOLS
Sequence submission refers to the submission of either protein or nucleic acid sequences to their respective databases. Sequence submission can be classified different ways depends on the source and sequence type.
Depending upon the source, it might be of two types namely web based submission tools and stand alone tools / softwares submission methods.
In web based (online) submission methods, sequence might be submitted through online submission methods like bankit (nucleotide sequence - ncbi), webin (nucleotide sequence - ebi) and spin (protein sequence - ebi).
In stand alone programs, sequence might be submitted through email after completing basic processing using softwares like sequin (protein and nucleotide sequences - ncbi).
Depending upon sequence nature different sequence methods might be used. For example in NCBI, depending upon different nature of sequences, different methods were used. They were listed in the following table.
Mainly three ways submission used on the basis of sequence nature, namely, simple sequence submission, genome sequence submission and other sequence submission types.
SAKURA:
The nucleotide sequence database created and maintained by the international collaboration of DDBJ/EMBL/GenBank is growing rapidly: the doubling time of the database has been steadily shortened and it is now only about a year. This fast growth of the database is largely attributable to the systematic genome projects, but the significant contribution of an increasing number of individual researchers around the world should not be underestimated. To make the data submission of the latter category easier and simpler, therefore, it is important to develop an efficient logistics system which facilitates faster data flow from submitters to the user community.
For this reason, we have developed a new data submission system based on the World Wide Web (WWW) and termed it SAKURA. The system has distinct advantages over the previous data submission by e-mail or floppy disk and the data acquisition process has become more reliable and efficient than before as described below.
(1) Expandability
SAKURA is made flexible to accommodate changes in the data specifications. For this purpose, the parser, page design, format, menu and language information are stored in separate resource _les independent of the computer programs that constitute SAKURA.
(2) Languages
All data entries must be prepared in English, but users can choose other languages for communication with the server during their submission sessions. At present, only Japanese or English are options. However, since SAKURA is capable of supporting multiple languages if appropriate language resource files are prepared, we plan to support, for example, Korean and Chinese in the near future.
(3) Pause and resume
SAKURA provides submitters with an option to 'pause and resume' his/her data submission session: the typed-in data will temporarily be held on the server for a period of up to a month, unless the submitter explicitly terminates the session. During this period, the submitter is able to retrieve the data from the server, modify and append them, if necessary. This capability will also enable submitters to copy and paste annotation lines when submitting multiple sets of data.
(4) Multiple entries
Consecutive accession numbers will be issued If multiple sets of data are received from a single submitter. The process will be automatically performed by the SAKURA system.
(5) Error check
Errors are more rigorously checked by Sakura than floppy disk or E-mail submissions. SAKURA classifies three types of errors, mandatory, illegal, or semantic, and will issue error messages. A 'mandatory' error message will be issued when any of the items that are absolutely required for data-processing are not supplied. An 'illegal' error message appears when the data includes illegal characters defined in the 'form' resource file. A 'semantic' error includes an erroneous biological description: for example, under the CDS feature identifier, the amino acid sequence that are experimentally determined and provided by the submitter must be identical to the one predicted from the DNA sequence. In addition, Sakura also issues warning messages whenever appropriate.
The first version of SAKURA was made available on the DDBJ WWW server in December 1995. The number of submissions using SAKURA has been steadily increasing since then and more than 70 % of the submissions were made with SAKURA in July 1995. This clearly indicates that an easy-to-use interface for data submission is indispensable for both submitters and the database. Currently, we are planning to convert the gateway scripts of the server into JAVA language so as to improve the user interface further.
Sakura is available at the following address: http://sakura.ddbj.nig.ac.jp. To get information regarding sequence submission contact sakura-admin@ddbj.nig.ac.jp. But unfortunately, submission through "SAKURA" was terminated on October 31, 2012 at 17:00(JST).
New submission page of DDBJ is http://ddbj.nig.ac.jp/submission/
We recommend you to use either one of the following internet browsers, because we tested the system only on these environments. Firefox, Chrome
Do not use [Back] button of web browser during submission.
You can resume the submission from the bookmark, if you bookmark the page.
Note: In order to suspend the submission except for "7.Annotation" page, you must click [Next] after you fill each input field.
We strongly recommend you to read Nucleotide Sequence Submission link http://www.ddbj.nig.ac.jp/submission-e.html before your submission
Check your sequences by DDBJ Vector Screening System (http://vector.ddbj.nig.ac.jp/top-e.html) to exclude vector sequences before submission.
You can see HELP file of the submission system (PDF) and illustrated instruction by clicking [Help] icon on each page.
If you have any questions on the usage of this system, send your question with URL of the submission page from Contact form. On the next page, select an item, "DDBJ Nucleotide Submission System".
Please configure your blocking tools against unwanted junk E-mails, NOT to block E-mails from DDBJ.
TPA data submission is also acceptable via this system. When you like to submit TPA data, select "constructed by using cited sequences" on sequence input page, "Sequences".
Eight steps of new submission steps in ddbj submission:
Enter contact person
Enter hold data
Enter submitter(s)
Enter reference
Enter nucleotide sequence
Select template that matches to annotation
7.
Annotation – text input field
7.
Annotation – link to the pages that explain error/warning
Completion of submission
Use BankIt if:
Use Sequin if:
BANKIT:
GenBank users may now use the World Wide Web (WWW) for submitting sequences to GenBank. The new submission tool - BankIt - provides a simple forms approach for submitting your sequence and descriptive information to GenBank. BankIt has been developed by GenBank in conjunction with its international collaborating databases (EMBL and DDBJ) and it is anticipated that EMBL and DDBJ will be offering similar services in the near future. Your submission will be submitted directly to GenBank and immediately forwarded for inclusion in the EMBL and DDBJ databases.
BankIt allows you to enter sequence information into a form, revise as necessary, and add biological annotation (e.g., coding regions, mRNA features). BankIt transforms your data into GenBank format for your review and when your record is completed, it can be submitted directly to GenBank. You have the option of adding information by using text boxes to describe in your own words the source of the sequence and its biological features. The GenBank annotation staff reviews the submitted textual information, incorporates it into the appropriate structured fields, and returns the record by e-mail for your review.
GenBank will promptly contact you by e-mail with an accession number. Once the testing phase is completed, BankIt will automatically issue accession numbers. BankIt has been tested with Netscape clients for Unix, Macs, and PCs. In addition, the Mosaic client for Unix, and the MacWeb client for Macs, have successfully been used.
You can access BankIt through the login page: http://www.ncbi.nlm.nih.gov/WebSub/?form=login&tool=genbank
If you have any questions on using BankIt, please contact the GenBank support staff at 'info@ ncbi.nlm.nih.gov'
Homepage for bankit

Bankit submission steps:
A BankIt submission involves seven easy steps:
SEQUIN:
Sequin is a stand-alone software tool developed by the NCBI for submitting and updating entries to the GenBank sequence database. It is capable of handling simple submissions that contain a single short mRNA sequence, and complex submissions containing long sequences, multiple annotations, gapped sequences, or phylogenetic and population studies. A single Sequin file should contain less than 10,000 sequences for maximum performance. Larger submissions should be made with tbl2asn. It also allows sequence editing and updating, and provides complex annotation capabilities. In addition, Sequin contains a number of built-in validation functions for enhanced quality assurance. The help information might be obtained from info@ncbi.nlm.nih.gov.
Sequin can display the initial record in a number of different formats. The record can be seen as it would appear in the GenBank, EMBL, or DDBJ databases. Sequences and certain annotations can also be viewed in a graphical format, permitting, for example, a schematic display of the locations of mRNAs and coding sequences along a genomic DNA sequence. If you have submitted a set of aligned sequences, the alignments can be displayed as well.
Sequin automatically performs a number of functions necessary for submission. For example, Sequin obtains the proper genetic code from the name of the organism and automatically determines coding region intervals on the nucleotide sequence by back-translation of the protein sequence. Researchers who submit large numbers of related sequences can make use of the fact that Sequin can also interpret the name of the organism, strain, and other biological source information directly from a line of data entered along with each nucleotide sequence. Sequin also allows the designation of groups of sequences as population, phylogentic, mutant, or environmental sets for display in the PopSet division of Entrez and the propagation of annotation from one member of the set to all others through an alignment.
A number of powerful sequence annotation tools have been integrated into Sequin. The ORF Finder identifies open reading frames within the sequence. The Sequence Editor allows basic editing and translation of nucleotide sequences. With the Update Sequence function, Sequin can import and align a replacement or overlapping sequence to the sequence in the record and propagate features between the two aligned sequences. In Network-Aware mode, Sequin integrates PubMed searching. Sequin also allows the propagation of features from one sequence in an aligned set to other sequences within the set.
Download sequin
Sequin 12.30 is currently available from the NCBI. Sequin runs on Macintosh, PC/Windows, and UNIX computers. The program itself, along with its on-line help documentation, is available by anonymous FTP (ftp://ftp.ncbi.nih.gov/sequin/).
Sequence submission using Sequin:
Sequin is organized into a series of forms for entering submitting authors, entering organism and sequences, entering information such as strain, gene, and protein names, viewing the complete submission, and editing and annotating the submission. The goal is to go quickly from raw sequence data to an assembled record that can be viewed, edited, and submitted to your database of choice.
Advance through the pages that make up each form by clicking on labeled folder tabs or the button. After the basic information forms have been completed and the sequence data imported, Sequin provides a complete view of your submission, in your choice of text or graphic format. At this point, any of the information fields can be easily modified by double-clicking on any area of the record, and additional biological annotations can be entered by selecting from a menu. Sequin has an on-screen file that is opened automatically when you start the program. Because it is context sensitive, the text will change and follow your steps as you progress through the program. A "Find" function is also provided.
Sequence submission using sequin achieved by following steps:
1. Prepare submission file before submission
Sequin normally expects to read sequence files in FASTA format. Population studies, phylogenetic studies, mutation studies, and environmental samples may be entered in either FASTA format, or in PHYLIP, NEXUS, MACAW, or FASTA+GAP formats if you are submitting an alignment. FASTA format is simply the raw sequence preceded by a definition line. The definition line begins with a > sign and is followed immediately by a name for the sequence (your own local identification code, or sequence ID) and a title. During the submission process, indexing staff at the database to which you are submitting will change your sequence ID to an Accession number. You can embed other important information in the title, and Sequin uses this information to construct a record. Specifically, you can enter organism and strain or clone information in the nucleotide definition line and gene and protein information in the protein definition line using name-value pairs surrounded by square brackets. Example: [organism=Drosophila melanogaster] [strain=Oregon R]
Some modifier names have restricted values or formats.
2. Start sequin program
Sequin's first window asks you to indicate the database to which the sequence will be submitted and prompts you to start a new project or continue with an existing one. Once you choose a database, Sequin will remember it in subsequent sessions. In general, each sequence submission should be entered as a separate project. However, segmented DNA sequences, gapped sequences, population studies, phylogenetic studies, and mutation studies should be submitted together as one project. To begin creating your submission, click the button.
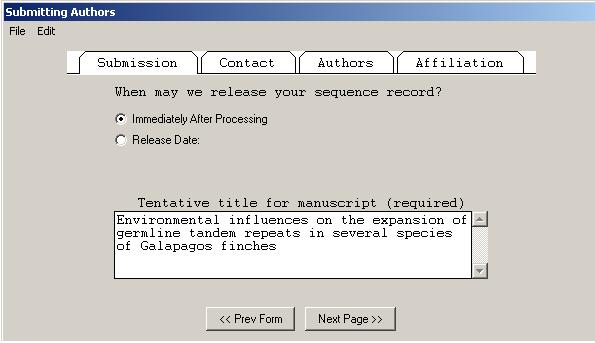
The pages in the Submitting Authors form ask you to provide the release date, a working title, names and contact information of submitting authors, and affiliation information. To create a personal template for use in future submissions, use the menu item after completing each page of this form.
The Submission page asks for a tentative title for a manuscript describing the sequence and will initially mark the manuscript as being unpublished. When the article is published, the database staff will update the sequence record with the new citation. This page also lets you indicate that a record should be held confidential by the database until a specified date, although the preferred policy is to release the record immediately into the public databases.
The Contact page asks for the name, phone number, and email address of the person responsible for making the submission. Database staff members will contact this person if there are any questions about the record. The Sfx (suffix) popup is used to enter personal name suffixes (e.g., Jr., Sr., or III), not a person's academic degrees (e.g., MD or PhD). Also, it is not necessary to type periods after initials.
In the Authors page, enter the names of the people who should get scientific credit for the sequence presented in this record. These will become the authors for the initial (unpublished) manuscript. Authors are entered in a spreadsheet. As soon as anything is typed in the last row, a new (blank) row is added below it. Use the tab key to move between fields. Tabbing from the last column automatically moves to the First Name column in the next row. The Affiliation page asks for the institutional affiliation of the primary author.
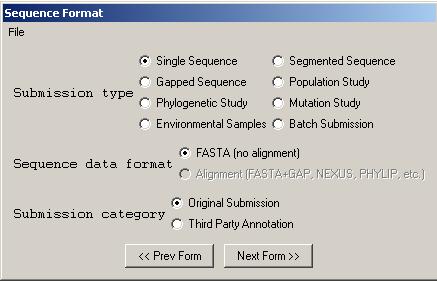
If you have sequence data from a single source, choose from one of the following submission types:
If you have a set of single sequences, segmented sequences, or gapped sequences or a mixture of these types of sequences, you will need to choose one of the following submission types:
If you have chosen , , , or for the submission type, you will only be able to select
If you have chosen one of the other submission types, you may import the sequences in FASTA format, or you may choose to import the sequences using an alignment file by selecting .
Choose if you have directly sequenced the nucleotide sequence in your laboratory. Choose if you have downloaded or assembled sequence from GenBank and modified it with your own annotations.
5. Organism and sequence form
The Organism and Sequences form has been enhanced with a number of Assistants that allow entry or editing of sequence and source information.
The Nucleotide page will have one of three appearances, based on whether you have chosen to import a single sequence, a set of sequences, or an alignment. To import a single sequence, click on and enter the name of the file that contains your FASTA sequence. In addition to importing from a file, sequences can also be read by pasting from the computer's "clipboard" using the menu item or by using the button. When the sequence file or alignment file import is complete, a box will appear showing the number of nucleotide segments imported, the total length in nucleotides of the sequences entered, and the sequence ID(s) you designated. The actual sequence data are not shown. If any of this information is missing or incorrect, check the file containing the sequence data for proper FASTA format, click on the button, then reimport the sequence(s).
If the imported nucleotide sequence or sequences or alignment have any problems, such as colliding local identifiers in a set or mismatched brackets in the definition line, an Assistant dialog appears to help correct the problems. Severe problems must be fixed before you can continue with the Sequin submission.
The second page of the Organism and Sequences form requests information regarding the scientific name of the organism from which the sequence was derived, if it was not already encoded in the nucleotide FASTA file. There are Assistants for manually adding organism name information or adding source qualifiers. Sequin has extracted the organism and strain names from the FASTA definition line in this example, eliminating the need to manually enter information in the Organism page.
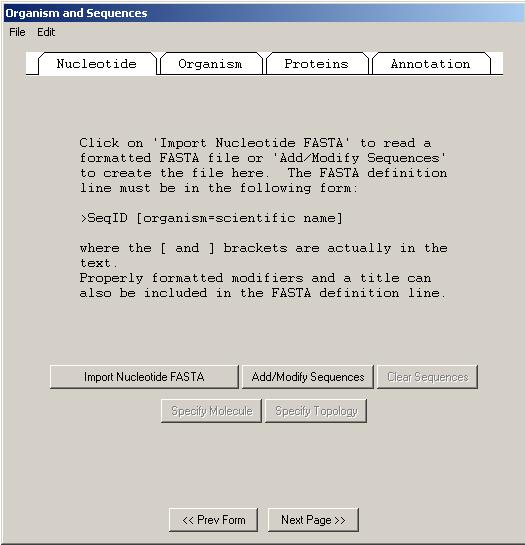
If your sequence or sequences encode one or more proteins, you can enter the sequences of the protein products in this page. To import the amino acid sequences, click on the Proteins folder tab and click on the button. You may import more than one file by clicking the button again after importing the first file.
The Annotation page allows you to add an rRNA or CDS feature to the entire length of all sequences in the set. In addition, you can add a title to any sequences that didn't obtain them from a FASTA definition line. It is much easier to add these in bulk at this step than to add individual rRNA or CDS features to each sequence after the record is constructed. The choice of "mRNA" or "gene" depends upon the molecule type (use "mRNA" for mRNA or cDNA, and "gene" for genomic DNA). Use "partial" for incomplete features. The proper organism name in a phylogenetic study can be added to the beginning of each title automatically by checking the box.
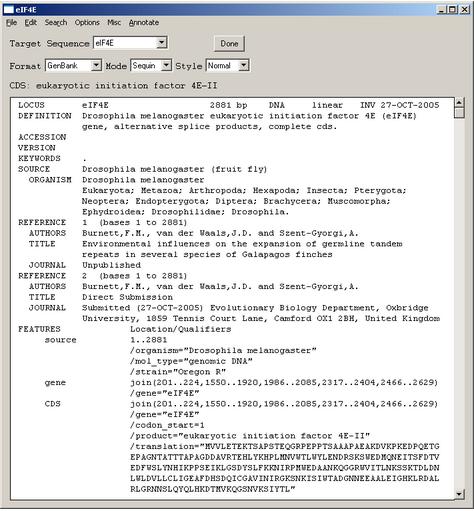
6. Viewing your submission
After you have completed importing the data files, Sequin will display your full submission information in the GenBank format. On the basis of the information provided in your DNA and amino acid sequence files, any coding regions will be automatically identified and annotated for you. The figure shows only the top portion of the GenBank record, but you can see the first of two coding region (CDS) features. The vertical bar to the left of the paragraph indicates that the CDS has been selected by clicking with the computer's mouse.
You may now make changes to the coding region, publication, source, and other features in the record by double clicking on the appropriate paragraphs in the GenBank display format. You may also use the menu item to compute a definition line for the annotated features in the record.
Two other viewing formats are available namely graphical format and sequence view. Reviewing your submission in Graphic format allows you to visually confirm expected location of exons, introns, and other features in multiple interval coding regions. Sequence view is a static version of the sequence and alignment editor. It shows the actual nucleotide sequence, with feature intervals annotated directly on the sequence.
7. Editing and annotating your submission
At this point, Sequin could process your entry based on what you have entered so far, and you could send it to your nucleotide database of choice (as set in the initial form). However, to optimize the usefulness of your entry for the scientific community, you may want to provide additional information to indicate biologically significant regions of the sequence. But first, save the entry so that if you make any unwanted changes during the editing process you can revert to the original copy. Additional information may be in the form of Descriptors or Features. Descriptors are annotations that apply to an entire sequence or set of sequences. They are used to remove redundant information in a record. Features are annotations that apply to a specific sequence interval.
Sequin provides two methods to modify your entry: (1) to edit existing information, double click on the text or graphic area you want to modify, and Sequin will display forms requesting needed information; or (2) to add new information, use the menu and select from the list of available annotations.
The menu item can make the appropriate titles once the record has been annotated with features. The general format for sequences containing coding region features is:
Genus species protein name (gene symbol) mRNA/gene, complete/partial cds.
Exceptional cases, where this automatic function is unable to generate a reasonable definition line, will be edited by the database staff to conform to the style conventions. The new definition line will replace any previous title, including that originally on the FASTA definition line.
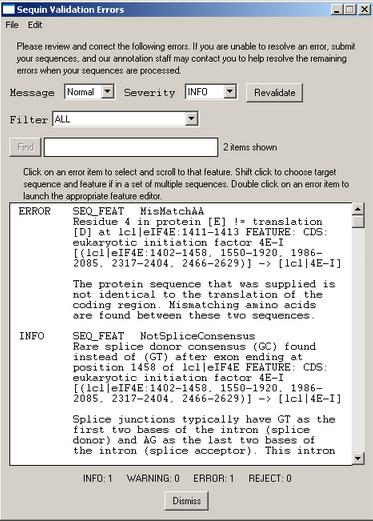
8. Record validation
Once you are satisfied that you have entered all the relevant information, save your file! Then select the menu item. You will either receive a message that the validation test succeeded or see a screen listing the validation errors and warnings. Just double click on an error item to launch the appropriate editor for making corrections. The validator includes checks for such things as missing organism information, incorrect coding region lengths, internal stop codons in coding regions, inconsistent genetic codes, mismatched amino acids, and non-consensus splice sites.
9. Submission
When the entry is properly formatted and error-free, click the button or select the menu item. You will be prompted to save your entry and email it to the database you selected. The address for GenBank is gb-sub@ncbi.nlm.nih.gov. The address for EMBL is datasubs@ebi.ac.uk. The address for DDBJ is ddbjsub@ddbj.nig.ac.jp.
Tbl2asn
The tbl2asn command line program is available via ftp and is designed as an alternative to the Sequin program for generating large single submissions (complete genomes) containing a great deal of annotation. It can also be used to generate a batch submission containing thousands of individual sequences.
SEQUENCE RETRIEVAL SYSTEM
ENTREZ:
The Entrez Global Query Cross-Database Search System is a powerful federated search engine, or web portal that allows users to search many discrete health sciences databases at the National Center for Biotechnology Information (NCBI) website. Entrez is NCBI’s primary text search and retrieval system that integrates the PubMed database of biomedical literature with 39 other literature and molecular databases including DNA and protein sequence, structure, gene, genome, genetic variation and gene expression. The NCBI is a part of the National Library of Medicine (NLM), which is itself a department of the National Institutes of Health (NIH), which in turn is a part of the United States Department of Health and Human Services. "Entrez" also happens to be the second person plural (or formal) form of the French verb "entrer (to enter)", meaning the invitation "Come in!".
Entrez Global Query is an integrated search and retrieval system that provides access to all databases simultaneously with a single query string and user interface. Entrez can efficiently retrieve related sequences, structures, and references. The Entrez system can provide views of gene and protein sequences and chromosome maps.
Features
The Entrez front page provides, by default, access to the global query. All databases indexed by Entrez can be searched via a single query string, supporting boolean operators and search term tags to limit parts of the search statement to particular fields. This returns a unified results page, that shows the number of hits for the search in each of the databases, which are also links to actual search results for that particular database.
Entrez also provides a similar interface for searching each particular database and for refining search results. The Limits feature allows the user to narrow a search a web forms interface. The History feature gives a numbered list of recently performed queries. Results of previous queries can be referred to by number and combined via boolean operators. Search results can be saved temporarily in a Clipboard. Users with a MyNCBI account can save queries indefinitely and also choose to have updates with new search results e-mailed for saved queries of most databases. It is widely used in the field of biotechnology to enhance the knowledge of students worldwide. It is a Life science search engine It is used in Bioinformatics.
The Entrez search interface features powerful options for constructing precise searches and managing results. Options include popular configurable Limits and preset filters to help focus on specific kinds of results, an Advanced Search interface that facilitates constructing more sophisticated queries. Specialized search fields are available for each database and can be browsed and selected in the Search Builder section of the Advanced Search interface. Other useful Entrez features include Search History with access to recent results and a Clipboard where search results can be saved temporarily. A My NCBI account increases the power of the system by providing even more flexibility. Most importantly Entrez integrates data with links within and between databases. Not only does this interconnectivity enhance navigation and allow search results to be quickly focused or expanded; but also, more importantly, these relationships often expose unexpected connections that can lead to scientific discoveries.

Entrez search
Nearly all search boxes that appear on the NCBI site access the Entrez system. The search box at the top of the NCBI homepage is a convenient place to begin Entrez searches. With the default All Databases selection, the results are presented on the Global Query page shown in Figure. This page lists the Entrez databases and the corresponding number of records found by the query in each database. The databases are organized into three sections on the Global Query page. The top section contains the literature databases: PubMed, PubMed Central, Books, OMIM and OMIA. The 26 molecular databases occupy the largest central section of the page. The bottom section hosts the accessory literature databases Journals, MeSH, and NLM Catalog. Of course, the Global Query page itself can be used to search all database by entering a simple search term or phrase in the Search across databases query box. Clicking on the number or the adjacent database name in Global Query retrieves the results in that database.
The search box on the NCBI homepage also has a pull-down list that allows selection of any of the individual databases. Alternatively, searches can be launched from the individual Entrez database pages. Many of the database homepages are linked directly to the NCBI homepage from the Popular Resource box in the upper right or from the lists in the footer area. All Entrez homepages are linked from the Resources list on the NCBI homepage. It is also easy to access the database homepages directly using the simplified addresses that are formed by adding the database name to that of the NCBI homepage. For example, the address for the gene database homepage is simply www.ncbi.nlm.nih.gov/gene. Searches launched from the database homepage allow for more precise search strategies tailored to the database.
Entrez databases
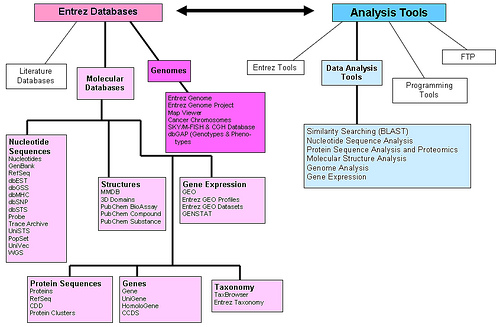
Entrez searching options:
Entrez queries can be single words, short phrases, sentences, database identifiers, gene symbols, or names … just about anything. Often simple searches can result in overwhelming numbers of results or even no results at all. There are a number of built-in Entrez features that can help in creating more effective queries. These include Boolean operators, query translation, and fielded searching using any of the indexed fields available for the database. Any of these can be used in manually writing and editing queries but are also incorporated into various aspects of the interface so that precise results are available without the need to write complex query statements. These aspects of the interface include limits, filters, and an Advanced Search page with a Search Builder and Search History that can be used to generate more sophisticated queries.
Using Boolean Operators
Boolean operators provide a way of generating precise queries that produce well-defined sets of results. The Boolean operators used in Entrez and how they work are as follows.
AND: Finds documents that contain terms on both sides of the operator terms, the intersection of both searches.
OR: Finds documents that contain either term, the union of both searches.
NOT: Finds documents that contain the term on the left but not the term on the right of the operator, the subtraction of the right hand search from the one on the left.
Entrez requires the Boolean operator AND to be entered in uppercase. This is not required in all databases for the other two operators, but it is simplest to enter all of them in uppercase:
promoters OR response elements NOT human AND mammals
Entrez processes all Boolean operators in a left-to-right sequence. Enclosing individual concepts in parentheses changes this priority. The terms inside the parentheses are processed first as a unit and then incorporated into the overall strategy. For example, in the following search statement, the union of response element and promoter results is generated first and then is intersected with the result of the g1p3 search.
g1p3 AND (response element OR promoter)
Display and send option
The Display Settings and Send to menus at the upper left and upper right of Entrez pages manage how records are displayed and stored or downloaded. The Display Settings menu has options for format, number of results per page, and sorting order. The available formats and sorting options vary depending on the database. The default format for multiple search results in Entrez is the Summary format that is consistent across databases. Single record default formats depend on the database. The default number of records displayed is 20 per page presented in the default (arbitrary) sorting order for the database. These default settings may be modified by setting personal Preferences in a My NCBI account.
The Send to menu has options for sending results to online storage in Collections in My NCBI, the NCBI Clipboard for the database, or to a local file. Additional options may be available depending on the database. When choosing the file option, the record format and sorting order can be specified. By default all Display Settings and Send to menu operations affect all records unless individual items are selected using the checkboxes at the left of the record title.
Acess
In addition to using the search engine forms to query the data in Entrez, NCBI provides the Entrez Programming Utilities (eUtils) for more direct access to query results. These are a set of eight server-side programs that provide a stable interface to the Entrez query and database system. The eUtils are accessed by posting specially formed URLs to the NCBI server, and parsing the XML response. There is also an eUtils SOAP interface.
SEQUENCE RETRIEVAL SYSTEM (SRS):
SRS (sequence retrieval system) is one of the most powerful data browsing/retrieval tools available. SRS provides rapid, easy and user friendly access to the large volumes of diverse and heterogeneous Life Science data stored in more than 400 internal and public domain databases. It can be used to browse the various biological sequence and literature databases the EBI has available. SRS is a powerful searching tool to retrieve sequences (and other types of data), and also to perform various operations on retrieved information (for example, you can align selected sequences using ClustalW2).
SRS is a homogeneous interface to over 80 biological databases that had been developed at the European Bioinformatics Institute (EBI) at Hinxton, UK. It includes databases of sequences, metabolic pathways, transcription factors, application results (like BLAST, SSEARCH, FASTA), protein 3-D structures, genomes, mappings, mutations, and locus speci_c mutations. The web page listing all the databases contains a link to a description page about the database including the date on which it was last updated. You select one or more of the databases to search before entering your query. After getting results you choose an alignment algorithm (like CLUSTALW, PHYLIP) enter parameters, and run it. The SRS is highly recommended for use.
The website is http://srs.ebi.ac.uk/.
The first page presents you with the menu, to start a one-off session click "Start". You are presented with the "TOP PAGE", where you can define which databank(s) to query. The EMBL database exists in two parts - EMBL and EMBLNEW. EMBL contains all entries present in the last database release, EMBLNEW - contains all newly created or updated entries since the last release. Database releases are produced in three months cycles, at time of building a new database release, contents of EMBLNEW are merged into EMBL data set. The date of the last database release can be found at the EMBL database web pages. To make sure that your query picks all possible entries, tick both "EMBL" and "EMBLNEW" checkboxes.
2. After selecting the databank(s), continue by either clicking "QUERY" bookmark or selecting "Standard" or "Extented" query forms.
3. On the "QUERY" page, enter details of your query. To choose the data fields you want to query, use the pull-down menu. On the "Standard" form one datafield per box can be selected.
Enter terms of
your query into the datafields
(examples below), and click on "Submit query".
You will be presented with a clickable list of entries (could be few pages long
depending on the type of query). To submit a new query, click again on
"QUERY" bookmark at the top of the page.
Query examples:
Difference between standard and Extended query forms
The "Extended query form" provides many more fields to construct your query (plus some types of queries are presented in a different form). For example, to query EST entries via the standard form, first choose field name "Division" and then type "est" into that field. In the "Extended" form tick the box "est" in "Division" section of the form. The “Extended" query form is especially useful for limiting the number of entries in the results.
SRS Concepts
SRS is designed to retrieve data directly from text files. From the beginning of the computer era text files in ASCII format have been widely accepted as a format to exchange information. This makes them portable to any computer system. While the paradigm of computers expanded from just performing calculations to complex data management, it became obvious that plain text format is not efficient enough for these purposes. But text files are still widely used to exchange and distribute information. In fact, formatted text files are the de-facto standard for biological databases like EMBL and SWISS-PROT. Self-descriptive XML format has advanced features but it is still just plain text.
The key feature of SRS is its unique object oriented design. It uses meta-data to define a class for a database entry object and rules for text-parsing methods, coupled with the entry attributes. For object definition and parsing rules SRS uses its own scripting language, Icarus, for which a debugger has been recently implemented. While RDBMS are highly advanced for data management, SRS has advantages as a retrieval system: First, it is much faster (10-100 times) than retrieving whole records from large databases with complex data schemas (like EMBL). Second, since it retrieves data directly from flatfiles it is less demanding in terms of storage space requirements than RDBMS tables. The average difference of 2-5 times is significant in the case of large databases as EMBL, which is about 28 Gb in flatfile format at present. Third, it is reasonably easy to integrate new data with basic retrieval capabilities and extend it further to a more sophisticated data schema.The integrating power of SRS benefits from sharing the definitions of conceptually equal attributes amongst different data sets. This enforces uniformity and allows multiple-database queries. Searchable links between databases and customizable data representation are original features of SRS.
SRS version 8 was released from Biowisdom. Biowisdom accessing srs link is http://bips.u-strasbg.fr/srs83/frontpage.do#. This server page used to access protein sequence and structure databases.
Availability
SRS6 is a licensed product of LION bioscience AG freely available for academics. The EBI SRS server is a free central resource for molecular biology data as well as a reference server for the latest developments in data integration.
Linking
Data becomes more valuable in the context of other data. Besides enriching the original data by providing html linking, one of the original features of SRS is the ability to define indexed links between databases. These links reflect equal values of named entry attributes in two databases. It could be a link from an explicitly defined reference in DR (data reference) records in SWISS-PROT or an implicit link from SWISS-PROT to the ENZYME database by a corresponding EC (Enzyme Commission) number in the protein description.
The links are bi-directional, operate on sets of entries, can be weighted and can be combined with logical operators (AND, OR and NOT). This is analogous to a table of relations in a relational database schema that allows querying of one table with conditions applied to others. The user can search not only the data contained in a particular database but also any conceptually related databases and then link to the desired data. Using the linking graph, SRS makes it possible to link databases that do not contain direct references to each other. Highly cross-linked data sets become a kind of domain knowledge base. This helps to perform queries like “give me all proteins that share InterPro domains with my protein” by linking from SWISSPROT to InterPro and back to SWISS-PROT, or “give me all eukaryotic proteins for which the promoter is further characterised” by selecting only entries linked to the EPD (Eukaryotic Promoter Database) from the current set.
Data warehousing
A recent extension to SRS is PRISMA. One of the hardest chores in maintaining an upto-date SRS server is the constant hunting for new database releases and updates. Typically, the nightly update of the EBI SRS server consists of more than 1000 processes. PRISMA is a set of programs designed to automate this process. It integrates the monitoring for new data sets on remote servers, downloading and indexing.
PRISMA can execute a user-defined number of parallel sessions in order to increase updating throughput and reduce the time it takes for users to be able to query the new data. Administratively, PRISMA combines parallel threads execution, automatic report generation with graphical diagrams, automated recovery and offline data processing, making it relatively simple and easy to quickly identify problems and take corrective actions.
Data analysis applications
The introduction of the biosequence object in SRS allows the integration of various sequence analysis tools such as FASTA or CLUSTALW. This integration allows treating the text output of these applications like any other database. This enables linking to other databanks and user-defined data representations. Up to now about a dozen applications are already integrated into SRS and many others are in the pipeline. Expanding in this direction SRS becomes not only a data retrieval system but also a data analysis application server. Recent advances in application integration include different levels of user control over application parameters, support for different UNIX queuing systems (LSF, CODINE, DQS, NQS) and parallel threading. There is now also support for ‘user-owned data’ (the user’s own sequences), which make SRS a more comprehensive research tool.
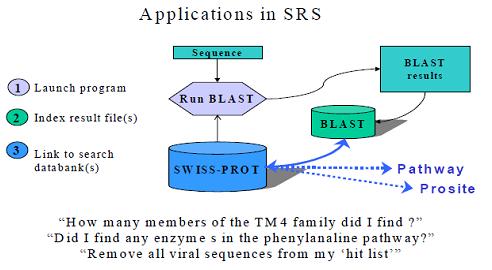
InterProScan
As an example of a data analysis application is InterProScan, which was recently implemented at the EBI. InterProScan is a wrapper on top of a set of applications for scanning protein sequences against InterPro member databases. Currently it is based on:
1. the FingerPRINTScan (Scordis P. et al. 1999) application that searches the PRINTS database for protein signatures;
2. ProfileScanner (pfscan) from the Pftools package for searching protein sequences against a collection of generalised profiles in PROSITE (http://www.isrec.isb-sib.ch/software/PFSCAN_form.html);
3. Ppsearch (Fuchs, R. 1994) for PROSITE pattern matching;
4. HMMPfam from the HMMER package (http://hmmer.wustl.edu/) or HMMS implemented on a Decypher machine from TimeLogic that scans sequences against the Pfam collection of protein domain HMMs (Hidden Markov Models).
InterProScan provides an efficient way to analyse protein sequences for known domains and functional sites by launching the applications in parallel, parsing their output and combining the results at the level of unified attributes into one representation with graphical visualisation of the matches.
Download viral protein using SRS-EBI
DBGET
DBGET is an integrated database retrieval system, developed at the University of Tokyo. It is provided access to 20 databases, one at a time. Having more limited options, the DBGET is less recommended than the two others.
Protein Sequences
The one-letter and three-letter abbreviation codes for amino acids for example, used in UniProtKB/Swiss-Prot are those adopted by the commission on Biochemical Nomenclature of the IUPAC-IUB and are as follows:
|
One-letter code |
Three-letter code |
Amino-acid name |
|
A |
Ala |
Alanine |
|
R |
Arg |
Arginine |
|
N |
Asn |
Asparagine |
|
D |
Asp |
Aspartic acid |
|
C |
Cys |
Cysteine |
|
Q |
Gln |
Glutamine |
|
E |
Glu |
Glutamic acid |
|
G |
Gly |
Glycine |
|
H |
His |
Histidine |
|
J |
Xle |
Leucine or Isoleucine |
|
L |
Leu |
Leucine |
|
I |
ILe |
Isoleucine |
|
K |
Lys |
Lysine |
|
M |
Met |
Methionine |
|
F |
Phe |
Phenylalanine |
|
P |
Pro |
Proline |
|
O |
Pyl |
Pyrrolysine |
|
U |
Sec |
Selenocysteine |
|
S |
Ser |
Serine |
|
T |
Thr |
Threonine |
|
W |
Trp |
Tryptophan |
|
Y |
Tyr |
Tyrosine |
|
V |
Val |
Valine |
|
B |
Asx |
Aspartic acid or Asparagine |
|
Z |
Glx |
Glutamic acid or Glutamine |
|
X |
Xaa |
Any amino acid |
Nucleotide Sequences
Nucleotide bases fall into two categories depending on the ring structure of the base. Purines (Adenine and Guanine) are two ring bases, pyrimidines (Cytosine and Thymine) are single ring bases. Mutations in DNA are changes in which one base is replaced by another. A mutation that conserves the ring number is called a transition (e.g., A -> G or C -> T) a mutation that changes the ring number are called transversions. (e.g. A -> C or A -> T and so on).
Nucleotide codes assigned by IUB
|
IUB |
Meaning |
Complement |
|
A |
A |
T |
|
C |
C |
G |
|
G |
G |
C |
|
T/U |
T |
A |
|
M |
A/C |
K |
|
R |
A/G |
Y |
|
W |
A/T |
W |
|
S |
C/G |
S |
|
Y |
C/T |
R |
|
K |
G/T |
M |
|
V |
A/C/G |
B |
|
H |
A/C/T |
D |
|
D |
A/G/T |
H |
|
B |
C/G/T |
V |
|
X/N |
A/C/G/T |
X |
|
. |
None |
. |
SEQUENCE FORMATS
Sequence formats are simply the way in which the amino acid or DNA sequence is recorded in a computer file. Different programs expect different formats, so it is important to understand various formats. One major difficulty encountered in running sequence analysis software is the use of differing sequence formats by different programs. These formats all are standard ASCII files, but they may differ in the presence of certain characters and words that indicate where different types of information and the sequence itself are to be found. The more commonly used sequence formats are discussed below.
Single sequence formats:
Genbank Format:
GenBank is the NIH genetic sequence database, an annotated collection of all publicly available DNA sequences. Each GenBank entry includes a concise description of the sequence, the scientific name and taxonomy of the source organism, and a table of features that identifies coding regions and other sites of biological significance, such as transcription units, sites of mutations or modifications, and repeats. Protein translations for coding regions are included in the feature table. Bibliographic references are included along with a link to the Medline unique identifier for all published sequences. Each sequence entry is composed of lines. Different types of lines, each with their own format, are used to record the various data that make up the entry.
Genbank format is also used for the databases in DDBJ. Protein sequence for the same format is known as Genpept at NCBI. One sequence in GenBank format starts with a line containing the word LOCUS and a number of annotation lines. The start of the sequence is marked by a line containing "ORIGIN" and the end of the sequence is marked by two slashes ("//").
An example sequence in GenBank format is:
LOCUS AB000263 368 bp mRNA linear PRI 05-FEB-1999
DEFINITION Homo sapiens mRNA for prepro cortistatin like peptide, complete
cds.
ACCESSION AB000263
ORIGIN
1 acaagatgcc attgtccccc ggcctcctgc tgctgctgct ctccggggcc acggccaccg
61 ctgccctgcc cctggagggt ggccccaccg gccgagacag cgagcatatg caggaagcgg
121 caggaataag gaaaagcagc ctcctgactt tcctcgcttg gtggtttgag tggacctccc
181 aggccagtgc cgggcccctc ataggagagg aagctcggga ggtggccagg cggcaggaag
241 gcgcaccccc ccagcaatcc gcgcgccggg acagaatgcc ctgcaggaac ttcttctgga
301 agaccttctc ctcctgcaaa taaaacctca cccatgaatg ctcacgcaag tttaattaca
361 gacctgaa
//
European Molecular Biology Laboratory (EMBL) Format
The European Molecular Biology Laboratory (EMBL) maintains DNA and protein sequence databases. As with GenBank entries, a large amount of information describing each sequence entry is given, including literature references, information about the function of the sequence, locations of mRNAs and coding regions, and positions of important mutations. This information is organized into fields, each with an identifier, shown as the first text on each line. These identifiers are abbreviated to two letters, e.g., RF for reference, and some identifiers may have additional subfields. The sequence entry is assumed by computer programs to lie between the identifiers “SEQUENCE” and “//” and includes numbers on each line to locate parts of the sequence visually. The sequence count or a checksum value for the sequence may be used by computer programs to make sure that the sequence is complete and accurate. For this reason, the sequence part of the entry should usually not be modified except with programs that also modify this count. This EMBL sequence format is very similar to the GenBank format. The main differences are in the use of the term ORIGIN in the GenBank format to indicate the start of sequence; also, the EMBL entry does not include the sequence of any translation products, which are shown instead as a different entry in the database. This sequence format often has to be changed for use with sequence analysis software. The meaning of each of these fields is as follows:
A complete and definitive description of the feature table is given here.
One sequence entry starts with an identifier line ("ID"), followed by further annotation lines. The start of the sequence is marked by a line starting with "SQ" and the end of the sequence is marked by two slashes ("//").
An example sequence in EMBL format is:
ID AB000263 standard; RNA; PRI; 368 BP.
XX
AC AB000263;
XX
DE Homo sapiens mRNA for prepro cortistatin like peptide, complete cds.
XX
SQ Sequence 368 BP;
acaagatgcc attgtccccc ggcctcctgc tgctgctgct ctccggggcc acggccaccg 60
ctgccctgcc cctggagggt ggccccaccg gccgagacag cgagcatatg caggaagcgg 120
caggaataag gaaaagcagc ctcctgactt tcctcgcttg gtggtttgag tggacctccc 180
aggccagtgc cgggcccctc ataggagagg aagctcggga ggtggccagg cggcaggaag 240
gcgcaccccc ccagcaatcc gcgcgccggg acagaatgcc ctgcaggaac ttcttctgga 300
agaccttctc ctcctgcaaa taaaacctca cccatgaatg ctcacgcaag tttaattaca 360
gacctgaa 368
//
UniProtKB/Swiss-Prot Format:
UniProtKB/Swiss-Prot is an annotated protein sequence database. The UniProtKB/Swiss-Prot protein knowledgebase consists of sequence entries. Sequence entries are composed of different line types, each with their own format. For standardisation purposes the format of UniProtKB/Swiss-Prot follows as closely as possible that of the EMBL Nucleotide Sequence Database. The entries in the UniProtKB/Swiss-Prot database are structured so as to be usable by human readers as well as by computer programs. The explanations, descriptions, classifications and other comments are in ordinary English. Wherever possible, symbols familiar to biochemists, protein chemists and molecular biologists are used. Each sequence entry is composed of lines. Different types of lines, each with their own format, are used to record the various data that make up the entry. The format of an entry in the SwissProt protein sequence database is very similar to the EMBL format, except that considerably more information about the physical and biochemical properties of the protein is provided.
An example sequence in UniProtKB/Swiss-Prot format is:
ID 100K_RAT STANDARD; PRT; 889 AA.
AC Q62671;
DE 100 kDa protein (EC 6.3.2.-).
SQ SEQUENCE 889 AA; 100370 MW; DD7E6C7A CRC32;
MMSARGDFLN YALSLMRSHN DEHSDVLPVL DVCSLKHVAY VFQALIYWIK AMNQQTTLDT
PQLERKRTRE LLELGIDNED SEHENDDDTS QSATLNDKDD ESLPAETGQN HPFFRRSDSM
TFLGCIPPNP FEVPLAEAIP LADQPHLLQP NARKEDLFGR PSQGLYSSSA GSGKCLVEVT
MDRNCLEVLP TKMSYAANLK NVMNMQNRQK KAGEDQSMLA EEADSSKPGP SAHDVAAQLK
SSLLAEIGLT ESEGPPLTSF RPQCSFMGMV ISHDMLLGRW RLSLELFGRV FMEDVGAEPG
SILTELGGFE VKESKFRREM EKLRNQQSRD LSLEVDRDRD LLIQQTMRQL NNHFGRRCAT
TPMAVHRVKV TFKDEPGEGS GVARSFYTAI AQAFLSNEKL PNLDCIQNAN KGTHTSLMQR
LRNRGERDRE REREREMRRS SGLRAGSRRD RDRDFRRQLS IDTRPFRPAS EGNPSDDPDP
LPAHRQALGE RLYPRVQAMQ PAFASKITGM LLELSPAQLL LLLASEDSLR ARVEEAMELI
VAHGRENGAD SILDLGLLDS SEKVQENRKR HGSSRSVVDM DLDDTDDGDD NAPLFYQPGK
RGFYTPRPGK NTEARLNCFR NIGRILGLCL LQNELCPITL NRHVIKVLLG RKVNWHDFAF
FDPVMYESLR QLILASQSSD ADAVFSAMDL AFAVDLCKEE GGGQVELIPN GVNIPVTPQN
VYEYVRKYAE HRMLVVAEQP LHAMRKGLLD VLPKNSLEDL TAEDFRLLVN GCGEVNVQML
ISFTSFNDES GENAEKLLQF KRWFWSIVER MSMTERQDLV YFWTSSPSLP ASEEGFQPMP
SITIRPPDDQ HLPTANTCIS RLYVPLYSSK QILKQKLLLA IKTKNFGFV
//
This format contains a single header line providing the sequence name, and optionally a description, followed by lines of sequence data. Sequences in FASTA formatted files are preceded by a line starting with a " >" symbol. The first word on this line is the name of the sequence. The rest of the line is a description of the sequence. The remaining lines contain the sequence itself, usually formated to 60 characters per line. Depending on the application blank lines in a FASTA file are ignored or treated as terminating the sequence. Depending on the application spaces or other non-sequence symbols (dashes, underscores, periods) in a sequence are either ignored or treated as gaps. The FASTA format is the one most often used by sequence analysis software. This format provides a very convenient way to copy just the sequence part from one window to another because there are no numbers or other non-sequence characters within the sequence. The FASTA sequence format is similar to the protein information resource (NBRF) format
An example sequence in FASTA format is:
>AB000263 Homo sapiens mRNA for prepro cortistatin like peptide, complete cds.|len=368
ACAAGATGCCATTGTCCCCCGGCCTCCTGCTGCTGCTGCTCTCCGGGGCCACGGCCACCGCTGCCCTGCC
CCTGGAGGGTGGCCCCACCGGCCGAGACAGCGAGCATATGCAGGAAGCGGCAGGAATAAGGAAAAGCAGC
CTCCTGACTTTCCTCGCTTGGTGGTTTGAGTGGACCTCCCAGGCCAGTGCCGGGCCCCTCATAGGAGAGG
AAGCTCGGGAGGTGGCCAGGCGGCAGGAAGGCGCACCCCCCCAGCAATCCGCGCGCCGGGACAGAATGCC
CTGCAGGAACTTCTTCTGGAAGACCTTCTCCTCCTGCAAATAAAACCTCACCCATGAATGCTCACGCAAG
TTTAATTACAGACCTGAA
National Biomedical Research Foundation/Protein Information Resource Sequence (NBRF/PIR) Format:
A sequence in PIR format consists of:
|
Sequence type |
Code |
|
Protein (complete) |
P1 |
|
Protein (fragment) |
F1 |
|
DNA (linear) |
DL |
|
DNA (circular) |
DC |
|
RNA (linear) |
RL |
|
RNA (circular) |
RC |
|
tRNA |
N3 |
|
other functional RNA |
N1 |
This sequence format, which is sometimes also called the PIR format, has been used by the National Biomedical Research Foundation/Protein Information Resource (NBRF) and also by other sequence analysis programs. Note that sequences retrieved from the PIR database on their Web site (http://www-nbrf.georgetown.edu) are not in this compact format, but in an expanded format with much more information about the sequence. There is also an essential second line with the full name of the sequence, a hyphen, then the species of origin. The NBRF format is similar to the FASTA sequence format but with significant differences.
An example sequence in NBRF/PIR format is:
>P1;CRAB_ANAPL
ALPHA CRYSTALLIN B CHAIN – Mus musculus
MDITIHNPLI RRPLFSWLAP SRIFDQIFGE HLQESELLPA SPSLSPFLMR
SPIFRMPSWL ETGLSEMRLE KDKFSVNLDV KHFSPEELKV KVLGDMVEIH
GKHEERQDEH GFIAREFNRK YRIPADVDPL TITSSLSLDG VLTVSAPRKQ
SDVPERSIPI TREEKPAIAG AQRK*
Stanford University/Intelligenetics Sequence Format:
A sequence file in IG format must begin with a semicolon (";"), a line with the sequence and the sequence itself terminated with the termination character '1' for linear or '2' for circular sequences. IG format Started by a molecular genetics group at Stanford University, and subsequently continued by a company, Intelligenetics, the IG format is similar to the PIR format, except that a semicolon is usually placed before the comment line.
An example sequence in IG format is:
;H.sapiens fau mRNA, 518 bases
HSFAU
ttcctctttctcgactccatcttcgcggtagctgggaccgccgttcagtc
gccaatatgcagctctttgtccgcgcccaggagctacacaccttcgaggt
gaccggccaggaaacggtcgcccagatcaaggctcatgtagcctcactgg
agggcattgccccggaagatcaagtcgtgctcctggcaggcgcgcccctg
gaggatgaggccactctgggccagtgcggggtggaggccctgactaccct
ggaagtagcaggccgcatgcttggaggtaaagttcatggttccctggccc
gtgctggaaaagtgagaggtcagactcctaaggtggccaaacaggagaag
aagaagaagaagacaggtcgggctaagcggcggatgcagtacaaccggcg
ctttgtcaacgttgtgcccacctttggcaagaagaagggccccaatgcca
actcttaagtcttttgtaattctggctttctctaataaaaaagccactta
gttcagtcaaaaaaaaaa1
Genetics Computer Group (GCG) Sequence Format:
The programs in the GCG suite of biological analysis software accept sequences in gcg format. A sequence file in GCG format contains exactly one sequence, begins with annotation lines and the start of the sequence is marked by a line ending with two dot ("..") characters. This line also contains the sequence identifier, the sequence length and a checksum. This format should only be used if the file was created with the GCG package. Earlier versions of the Genetics Computer Group (GCG) programs require a unique sequence format and include programs that convert other sequence formats into GCG format.
Later versions of GCG accept several sequence formats.
An example sequence in GCG format is:
!!NA_SEQUENCE 1.0
H.sapiens fau mRNA
HSFAU Length: 518 Type: N Check: 2981 ..
1 ttcctctttc tcgactccat cttcgcggta gctgggaccg ccgttcagtc
51 gccaatatgc agctctttgt ccgcgcccag gagctacaca ccttcgaggt
101 gaccggccag gaaacggtcg cccagatcaa ggctcatgta gcctcactgg
151 agggcattgc cccggaagat caagtcgtgc tcctggcagg cgcgcccctg
201 gaggatgagg ccactctggg ccagtgcggg gtggaggccc tgactaccct
251 ggaagtagca ggccgcatgc ttggaggtaa agttcatggt tccctggccc
301 gtgctggaaa agtgagaggt cagactccta aggtggccaa acaggagaag
351 aagaagaaga agacaggtcg ggctaagcgg cggatgcagt acaaccggcg
401 ctttgtcaac gttgtgccca cctttggcaa gaagaagggc cccaatgcca
451 actcttaagt cttttgtaat tctggctttc tctaataaaa aagccactta
501 gttcagtcaa aaaaaaaa
The first line starts with the text ENTRY". The end of a sequence is delineated by "///". The "SEQUENCE" line specifies the beginning of the sequence lines (starting on the next line), and no sequence is assumed to appear in the entry if the "SEQUENCE" line is missing.
An example sequence in NBRF/PIR Codata format is:
ENTRY IXI_234
SEQUENCE
5 10 15 20 25 30
1 T S P A S I R P P A G P S S R P A M V S S R R T R P S P P G
31 P R R P T G R P C C S A A P R R P Q A T G G W K T C S G T C
61 T T S T S T R H R G R S G W S A R T T T A A C L R A S R K S
91 M R A A C S R S A G S R P N R F A P T L M S S C I T S T T G
121 P P A W A G D R S H E
///
Plain/ASCII.Staden Sequence Format:
A sequence in plain format may contain only IUPAC characters and spaces (no numbers!). A file in plain sequence format may only contain one sequence, while most other formats accept several sequences in one file. This sequence format is a computer file that includes only the sequence with no other accessory information. This particular format is used by the Staden Sequence Analysis programs (http://www/.mrc-lmb.com.ac.uk/pubseq) produced by Roger Staden at Cambridge University. The sequence must be further formatted to be used for most sequence analysis programs. Staden formatted sequence files contain the sequence and nothing else.
An example sequence in plain format is:
ACAAGATGCCATTGTCCCCCGGCCTCCTGCTGCTGCTGCTCTCCGGGGCCACGGCCACCGCTGCCCTGCC
CCTGGAGGGTGGCCCCACCGGCCGAGACAGCGAGCATATGCAGGAAGCGGCAGGAATAAGGAAAAGCAGC
CTCCTGACTTTCCTCGCTTGGTGGTTTGAGTGGACCTCCCAGGCCAGTGCCGGGCCCCTCATAGGAGAGG
AAGCTCGGGAGGTGGCCAGGCGGCAGGAAGGCGCACCCCCCCAGCAATCCGCGCGCCGGGACAGAATGCC
CTGCAGGAACTTCTTCTGGAAGACCTTCTCCTCCTGCAAATAAAACCTCACCCATGAATGCTCACGCAAG
TTTAATTACAGACCTGAA
Abstract Syntax Notation (ASN.1) Sequence Format:
Abstract Syntax Notation (ASN.1) is a formal data description language that has been developed by the computer industry. ASN.1 (http://www-sop.inria.fr/rodeo/personnel/hoschka/asn1.html; NCBI 1993) has been adopted by the National Center for Biotechnology Information (NCBI) to encode data such as sequences, maps, taxonomic information, molecular structures, and bibliographic information. These data sets may then be easily connected and accessed by computers. The ASN.1 sequence format is a highly structured and detailed format especially designed for computer access to the data. All the information found in other forms of sequence storage, e.g., the GenBank format, is present. For example, sequences can be retrieved in this format by ENTREZ. However, the information is much more difficult to read by eye than a GenBank formatted sequence. One would normally not need to use the ASN.1 format except when running a computer program that uses this format as input.
An example sequence in ASN.1 format is:
seq {
id { local id 1 },
descr { title "" },
inst {
repr raw, mol aa, length 131, topology linear,
{
seq-data
iupacaa "TSPASIRPPAGPSSRPAMVSSRRTRPSPPGPRRPTGRPCCSAAPRRPQATGGWKTCSGTCT
TSTSTRHRGRSGWSARTTTAACLRASRKSMRAACSRSAGSRPNRFAPTLMSSCITSTTGPPAWAGDRSHE"
} }
Genetic Data Environment (GDE) Sequence Format:
Genetic Data Environment (GDE) format is used by a sequence analysis system called the Genetic Data Environment, which was designed by Steven Smith and collaborators (Smith et al. 1994) around a multiple sequence alignment editor that runs on UNIX machines. The GDE features are incorporated into the SEQLAB interface of the GCG software, version 9. GDE format is a tagged-field format similar to ASN.1 that is used for storing all available information about a sequence, including residue color. GDE format is a tagged field format used for storing all available information about a sequence. The format matches very closely the GDE internal structures for sequence data. The format consists of text records starting and ending with braces ('{}'). Between the open and close braces are several tagged field lines specifying different pieces of information about a given sequence. The tag values can be wrapped with double quote characters ('""') as needed. If quotes are not used, the first white space delimited string is taken as the value. Any fields that are not specified are assumed to be the default values. Offsets can be negative as well as positive. Genbank entries written out in this format will have all (") converted to ('), and all ({}) converted to ([]) to avoid confusion in the parser. Leading and trailing gaps are removed prior to writing each sequence. This format is deliberately verbose in order to be simple to duplicate.
An example sequence in GDE format is:
{
name "Short name for sequence"
longname "Long (more descriptive) name for sequence"
sequence-ID "Unique ID number"
creation-date "mm/dd/yy hh:mm:ss"
direction [-1|1]
strandedness [1|2]
type [DNA|RNA||PROTEIN|TEXT|MASK]
offset (-999999,999999)
group-ID (0,999)
creator "Author's name"
descrip "Verbose description"
comments "Lines of comments that can be fairly arbitrary text about a
sequence. Return characters are allowed, but no internal double quotes
or brace characters. Remember to close with a double quote"
sequence "gctagctagctagctagctcttagctgtagtcgtagctgatgctagct
gatgctagctagctagctagctgatcgatgctagctgatcgtagctgacg
gactgatgctagctagctagctagctgtctagtgtcgtagtgcttattgc"
}
Rich sequence format (RSF) Format:
RSF means rich sequence format and it is created by the Editor in SeqLab. The format is recognised by the word !!RICH_SEQUENCE at the beginning of the file. In addition to the sequence data, each sequence can be annotated with descriptive sequence information such as:
One way to create a file in rsf format is to use GCG's NetFetch program. This program will download the appropriate file from NCBI and save the result in RSF format.
An example sequence in RSF format is:
!!RICH_SEQUENCE 1.0
..
{
name chkhba
type DNA
longname chkhba
checksum 980
creation-date 4/15/98 16:42:47
strand 1
sequence
ACACAGAGGTGCAACCATGGTGCTGTCCGCTGCTGACAAGAACAACGTCAAGGGCATCTT
CACCAAAATCGCCGGCCATGCTGAGGAGTATGGCGCCGAGACCTTGGAAAGGATGTTCAC
CACCTACCCCCCAACCAAGACCTACTTCCCCCACTTCGATCTGTCACACGGCTCCGCTCA
...
}
MULTIPLE SEQUENCE FORMATS
Unaligned multiple sequence formats
Some sequence formats can hold multiple sequences in one file. The details of how many sequences are held in one file differs between formats, but they either allows many sequences to be concatenated one after the other, or they hold the sequences together in some sort of aligned set of sequences. Other formats, such as gcg, plain and staden formats can only hold one sequence per file. An attempt to concatenate several sequences in one file leaves the results as a mess that makes it impossible to decide where the sequences start and end or what is annotation and what is sequence. Unaligned formats are used as input sequence file format for multiple sequence alignment programs. They are prepared by simply copying multiple sequences one by one in a text file. They are as follows:
1. Fasta/Pearson format
>seq1
agctagct agct agct
>seq2
aactaact aact aact
2. Intelligenetics format
;seq1, 16 bases, 2688 checksum.
seq1
agctagctagctagct1
;seq2, 16 bases, 25C8 checksum.
seq2
aactaactaactaact1
3. GenBank format
LOCUS seq1 16 bp
DEFINITION seq1, 16 bases, 2688 checksum.
ORIGIN
1 agctagctag ctagct
//
LOCUS seq2 16 bp
DEFINITION seq2, 16 bases, 25C8 checksum.
ORIGIN
1 aactaactaa ctaact
//
4. NBRF format
>DL;seq1
seq1, 16 bases, 2688 checksum.
agctagctag ctagct*
>DL;seq2
seq2, 16 bases, 25C8 checksum.
aactaactaa ctaact*
5. EMBL format
ID seq1
DE seq1, 16 bases, 2688 checksum.
SQ 16 BP
agctagctag ctagct
//
ID seq2
DE seq2, 16 bases, 25C8 checksum.
SQ 16 BP
aactaactaa ctaact
//
6. GCG format
seq1
seq1 Length: 16 Check: 9864 ..
1 agctagctag ctagct
seq2
seq2 Length: 16 Check: 9672 ..
1 aactaactaa ctaact
7. Format for the Macintosh sequence analysis program DNA Strider
; ### from DNA Strider ;-)
; DNA sequence seq1, 16 bases, 2688 checksum.
;
agctagctagctagct
//
; ### from DNA Strider ;-)
; DNA sequence seq2, 16 bases, 25C8 checksum.
;
aactaactaactaact
//
8. Format for phylogenetic analysis programs of Walter Fitch
seq1, 16 bases, 2688 checksum.
agc tag cta gct agc t
seq2, 16 bases, 25C8 checksum.
aac taa cta act aac t
9. Format for phylogenetic analysis programs PHYLIP of J. Felsenstein v 3.3 and 3.4.
2 16
seq1 agctagctag ctagct
seq2 aactaactaa ctaact
10. Protein International Resource PIR/CODATA format
\\\
ENTRY seq1
TITLE seq1, 16 bases, 2688 checksum.
SEQUENCE
5 10 15 20
25 30
1 a g c t a g c t a g c t a g c t
///
ENTRY seq2
TITLE seq2, 16 bases, 25C8 checksum.
SEQUENCE
5 10 15 20
25 30
1 a a c t a a c t a a c t a a c t
///
ALIGNED MULTIPLE SEQUENCE FORMATS
ALN/ClustalW2 format:
ALN format was
originated in the alignment program ClustalW2. The file starts with word "CLUSTAL"
and then some information about which clustal program was run and the version of
clustal used.
e.g. "CLUSTAL W (2.1) multiple sequence alignment"
The type of clustal program is "W" and the version is 2.1.
The alignment is written in blocks of 60 residues.
Every block starts with the sequence names, obtained from the input sequence,
and a count of the total number of residues is shown at the end of the line.
The information about which residues match is shown below each block of
residues:
"*"
means that the residues or nucleotides in that column are identical in all
sequences in the alignment.
":" means that conserved substitutions have been observed.
"." means that semi-conserved substitutions are
observed.
An example is shown below.
CLUSTAL W 2.1 multiple sequence alignment
FOSB_MOUSE ITTSQDLQWLVQPTLISSMAQSQGQPLASQPPAVDPYDMPGTSYSTPGLSAYSTGGASGS 60
FOSB_HUMAN ITTSQDLQWLVQPTLISSMAQSQGQPLASQPPVVDPYDMPGTSYSTPGMSGYSSGGASGS 60
********************************.***************:*.**:******
Phylip format:
The first line of the input file contains the number of species and the number of characters separated by blanks. The information for each species follows, starting with a ten-character species name (which can include punctuation marks and blanks), and continuing with the characters for that species. Phylip format files can be interleaved, as in the example below, or sequential. An example phylip format file:
4 131
IXI_234 TSPASIRPPA GPSSRPAMVS SRRTRPSPPG PRRPTGRPCC SAAPRRPQAT
IXI_235 TSPASIRPPA GPSSR----- ----RPSPPG PRRPTGRPCC SAAPRRPQAT
IXI_236 TSPASIRPPA GPSSRPAMVS SR--RPSPPP PRRPPGRPCC SAAPPRPQAT
IXI_237 TSPASLRPPA GPSSRPAMVS SRR-RPSPPG PRRPT----C SAAPRRPQAT
GGWKTCSGTC TTSTSTRHRG RSGWSARTTT AACLRASRKS MRAACSRSAG
GGWKTCSGTC TTSTSTRHRG RSGW------ ----RASRKS MRAACSRSAG
GGWKTCSGTC TTSTSTRHRG RSGWSARTTT AACLRASRKS MRAACSR--G
GGYKTCSGTC TTSTSTRHRG RSGYSARTTT AACLRASRKS MRAACSR--G
SRPNRFAPTL MSSCITSTTG PPAWAGDRSH E
SRPNRFAPTL MSSCITSTTG PPAWAGDRSH E
SRPPRFAPPL MSSCITSTTG PPPPAGDRSH E
SRPNRFAPTL MSSCLTSTTG PPAYAGDRSH E
GCG/MSF Format:
The file may begin with as many lines of comment or description as required. The comments are terminated with a line starting with two slashes. The first mandatory line that is recognised as part of the MSF file is the line containing the text "MSF:", this line also includes the sequence length, type and date plus an internal check sum value. The next line is a mandatory blank line inserted before the sequence names. There then follows one line per sequence describing the sequence name, length, checksum and a weight value. Only one name per line is allowed; the qualifier "Name: " is followed by the sequence name. Names are restricted to 10 characters or less. Extra characters, between the sequence names and "Len: " are acceptable if they contain no blank characters. Another blank line is added followed by a line starting with two slashes "//" , this indicates the end of the name list. There then follows another blank line. Sequences are interleaved on separate lines with gaps represented by periods. Each sequence line starts with the sequence name which is separated from the aligned sequence residues by white space.
MSF: 510 Type: P Check: 7736 ..
Name: ACHE_BOVIN oo Len: 510 Check: 7842 Weight: 16.0
Name: ACHE_HUMAN oo Len: 510 Check: 8553 Weight: 17.8
Name: ACHE_MOUSE oo Len: 510 Check: 229 Weight: 12.5
Name: ACHE_RAT oo Len: 510 Check: 8410 Weight: 14.2
Name: ACHE_XENLA oo Len: 510 Check: 2702 Weight: 39.2
//
ACHE_BOVIN MAGALLCALL LLQLLGRGEG KNEELRLYHY LFDTYDPGRR PVQEPEDTVT
ACHE_HUMAN MARAPLGVLL LLGLLGRGVG KNEELRLYHH LFNNYDPGSR PVREPEDTVT
ACHE_MOUSE MAGALLGALL LLTLFGRSQG KNEELSLYHH LFDNYDPECR PVRRPEDTVT
ACHE_RAT MTMALLGTLL LLALFGRSQG KNEELSLYHH LFDNYDPECR PVRRPEDTVT
ACHE_XENLA MESGVRILSL LILLHNSLAS ESEESRLIKH LFTSYDQKAR PSKGLDDVVP
ACHE_BOVIN ISLKVTLTNL ISLNEKEETL TTSVWIGIDW QDYRLNYSKG DFGGVETLRV
ACHE_HUMAN ISLKVTLTNL ISLNEKEETL TTSVWIGIDW QDYRLNYSKD DFGGIETLRV
ACHE_MOUSE ITLKVTLTNL ISLNEKEETL TTSVWIGIDW HDYRLNYSKD DFAGVGILRV
ACHE_RAT ITLKVTLTNL ISLNEKEETL TTSVWIGIEW QDYRLNFSKD DFAGVEILRV
ACHE_XENLA VTLKLTLTNL IDLNEKEETL TTNVWVQIAW NDDRLVWNVT DYGGIGFVPV
AMPS Block file format:
The first part of a block-file contains the identifier codes of the sequences that are to follow. Each code is prefixed by the > symbol, codes must not contain spaces.
e.g.
>HAHU
>Trypsin
>A0046
>Seq1
etc.
The number of ">" symbols is read in the beginning of the file until a * symbol
is found. The * signals the beginning of the multiple alignment which is stored
VERTICALLY, thus columns are individual sequences, whilst rows are aligned
positions. The * symbol must lie over the first sequence. A further star in the
same column signals the end of the alignment. Software then uses the number of
">" symbols at the beginning of the file to work out how many columns to read
from the * position. It is therefore important that the only ">" symbols in the
file are those that define the identifiers, and the only symbols are those
defining the start and end of the multiple alignment. A simple, small block-file
is shown below.
>Seq_1
>A0231
>HAHU
>Four_Alpha
>Globin
>GLobin_C
*
ARNDLQ
AAAAAA
PPPPPP
PP PPP
WW WWW
LLLLLL
IIVVLL
*
Nexus format:
Format used by
phylogenetic analysis program PAUP (phylogenetic analysis using parsimony). ntax
is number of taxa, nchar is the length of the alignment, and interleave allows
the alignment to be shown in readable blocks. The other terms describe the type
of sequence and the character used to indicate gaps.
Command inside
square brackets [ and ] are ignored (comment). The NEXUS file format is
comprised a "blocks" such as the taxa block, data block, sets block, trees
block, PAUP block and MrBayes block, to name a few. Each block starts with
begin <block
name>;
and ends with
end;.
#NEXUS
Begin data;
Dimensions ntax=4 nchar=15;
Format datatype=dna symbols="ACTG" missing=? gap=-;
Matrix
Species1 atgctagctagctcg
Species2 atgctagctag-tag
Species3 atgttagctag-tgg
Species4 atgttagctag-tag
;
End;
Selex format:
The Selex format used by hidden Markov program HMMER by Sean Eddy has been used to keep track of the alignment of small RNA molecules. Each line contains a name, followed by the aligned sequence. A space, dash, underscore, or period denotes a gap. Long alignments are split into multiple blocks and interleaved or separated by blank lines. The number of sequences, their order, and their names must be the same in every block, and every sequence must be represented even though there are no residues present.
# seq1
# seq2
# seq3
seq1 ACGACGACGACG.
seq2 ..GGGAAAGG.GA
seq3 UUU..AAAUUU.A
seq1 ..ACG
seq2 AAGGG
seq3 AA...UUU
Pfam/Stockholm format:
The "Pfam/Stockholm"
format is a system for marking up features in a multiple alignment. These
mark-up annotations are preceded by a 'magic' label, of which there are four
types.
Header
The first line in the file must contain a format and version identifier,
currently:
#
STOCKHOLM 1.0
The sequence alignment
< seqname> <aligned sequence>
< seqname> <aligned sequence>
< seqname> <aligned sequence>
.
.
//
<seqname>
stands for "sequence name", typically in the form "name/start-end" or just
"name".
The "//" line indicates the end of the alignment.
Sequence letters may include any characters except whitespace. Gaps may be
indicated by "." or "-".
Wrap-around alignments are allowed in principle, mainly for historical reasons,
but are not used in e.g. Pfam. Wrapped alignments are discouraged since they are
much harder to parse.
The alignment mark-up
Mark-up lines may include any characters except whitespace. Use underscore ("_")
instead of space.
#=GF <feature>
<Generic per-File annotation, free text>
#=GC <feature> <Generic per-Column annotation, exactly 1 char per column>
#=GS <seqname> <feature> <Generic per-Sequence annotation, free text>
#=GR <seqname> <feature> <Generic per-Sequence AND per-Column markup, exactly 1
char per column>
Example:
# STOCKHOLM 1.0
#=GF ID CBS
#=GF AC PF00571
#=GF DE CBS domain
#=GF AU Bateman A
#=GF CC CBS domains are small intracellular modules mostly found
#=GF CC in 2 or four copies within a protein.
#=GF SQ 67
#=GS O31698/18-71 AC O31698
#=GS O83071/192-246 AC O83071
#=GS O83071/259-312 AC O83071
#=GS O31698/88-139 AC O31698
#=GS O31698/88-139 OS Bacillus subtilis
O83071/192-246 MTCRAQLIAVPRASSLAE..AIACAQKM....RVSRVPVYERS
#=GR O83071/192-246 SA 999887756453524252..55152525....36463774777
O83071/259-312 MQHVSAPVFVFECTRLAY..VQHKLRAH....SRAVAIVLDEY
#=GR O83071/259-312 SS CCCCCHHHHHHHHHHHHH..EEEEEEEE....EEEEEEEEEEE
O31698/18-71 MIEADKVAHVQVGNNLEH..ALLVLTKT....GYTAIPVLDPS
#=GR O31698/18-71 SS CCCHHHHHHHHHHHHHHH..EEEEEEEE....EEEEEEEEHHH
O31698/88-139 EVMLTDIPRLHINDPIMK..GFGMVINN......GFVCVENDE
#=GR O31698/88-139 SS CCCCCCCHHHHHHHHHHH..HEEEEEEE....EEEEEEEEEEH
#=GC SS_cons CCCCCHHHHHHHHHHHHH..EEEEEEEE....EEEEEEEEEEH
O31699/88-139 EVMLTDIPRLHINDPIMK..GFGMVINN......GFVCVENDE
#=GR O31699/88-139 AS ________________*__________________________
#=GR_O31699/88-139_IN ____________1______________2__________0____
//
The block multiple sequence alignment format:
Identification starts contain a short identifier for the group of sequences from which the block was made and often is the original Prosite group ID. The identifier is terminated by a semicolon, and “BLOCK” indicates the entry type. AC contains the block number, a seven-character group number for sequences from which the block was made, followed by a letter (A–Z) indicating the order of the block in the sequences. The block number is a 5-digit number preceded by BL (BLOCKS database) or PR (PRINTS database). min, max is the minimum, maximum number of amino acids from the previous block or from the sequence start. DE describes sequences from which the block was made. BL contains information about the block: xxx is the amino acids in the spaced triplet found by MOTIF upon which the block is based. w is the width of the sequence segments (columns) in the block. s is the number of sequence segments (rows) in the block. Other values (n1, n2) describe statistical features of the block. Sequence id is a list of sequences. Each sequence line contains a sequence identifier, the offset from the beginning of the sequence to the block in parentheses, the sequence segment, and a weight for the segment.
Example:
ID short_identifier; BLOCK
AC block_number; distance from previous block = (min,max)
DE description
BL xxx motif; width=w; seqs=s; 99.5%=n1; strength=n2
sequence_id (offset) sequence_segment sequence_weight.
//
ID GLU_CARBOXYLATION; BLOCK
AC BL00011; distance from previous block=(1,64)
DE Vitamin K-dependent carboxylation domain proteins.
BL ECA motif; width=40; seqs=34; 99.5%=1833; strength=1412
FA10_BOVIN ( 45) LEEVKQGNLERECLEEACSLEEAREVFEDAEQTDEFWSKY 31
FA10_CHICK ( 45) LEEMKQGNIERECNEERCSKEEAREAFEDNEKTEEFWNIY 46
FA10_HUMAN ( 45) LEEMKKGHLERECMEETCSYEEAREVFEDSDKTNEFWNKY 33
FA7_BOVIN ( 5) LEELLPGSLERECREELCSFEEAHEIFRNEERTRQFWVSY 57
FA7_HUMAN ( 65) LEELRPGSLERECKEEQCSFEEAREIFKDAERTKLFWISY 42
OSTC_CHICK ( 6) SGVAGAPPNPIEAQREVCELSPDCNELADELGFQEAYQRR 94
//
Known biosequence format Extensions
|
ID |
Name |
Suffix |
|
1 |
IG|Stanford |
.ig |
|
2 |
GenBank|GB |
.gb |
|
3 |
NBRF |
.nbrf |
|
4 |
EMBL |
.embl |
|
5 |
GCG |
.gcg |
|
6 |
DNAStrider |
.strider |
|
7 |
Fitch |
.fitch |
|
8 |
Pearson|FASTA |
.fasta |
|
9 |
Zuker |
.zuker |
|
10 |
Olsen |
.olsen |
|
11 |
Phylip3.2 |
.phylip2 |
|
12 |
Phylip|Phylip4 |
.phylip |
|
13 |
Plain|Raw |
.seq |
|
14 |
PIR|CODATA |
.pir |
|
15 |
MSF |
.msf |
|
16 |
PAUP|NEXUS |
.nexus |
|
17 |
Pretty |
.pretty |
|
18 |
XML |
.xml |
|
19 |
BLAST |
.blast |
|
20 |
SCF |
.scf |
|
21 |
ASN.1 |
.asn |
SEQUENCE FILE FORMAT CONVERTERS
READSEQ:
READSEQ is an extremely useful sequence formatting program developed by D. G. Gilbert at Indiana University, Bloomington (gilbertd_bio.indiana.edu). READSEQ can recognize a DNA or protein sequence file in any of the formats shown in Table, identify the format, and write a new file with an alternative format. Some of these formats are used for special types of analyses such as multiple sequence alignment and phylogenetic analysis. READSEQ may be reached at the Baylor College of Medicine site at http://dot.imgen.bcm.tmc.edu:9331/seq-util/readseq.html and also by anonymous FTP from ftp.bio.indiana.edu/molbio/readseq or ftp.bioindiana.edu/molbio/mac to obtain the appropriate files. Data files that have multiple sequences, such as those required for multiple sequence alignment and phylogenetic analysis using parsimony (PAUP), are also converted. Examples of the types of files produced are shown in Table 2.4. Options to reverse-complement and to remove gaps from sequences are included.
Readseq was written originally around 1989 a component of a sequence analysis program, in Pascal, but when I added a small, simple command-line interface, it took on a life of its own as a conversion program for bioinformatics. It's main contribution to bioinformatics is it takes on the job of guessing what your input biosequence data format is, and converting it to what your software knows how to handle.
It was converted to a C program in early 1990's and after an update in 1993, remained as is for several years, as I wrote around it and moved thru C++ then on to Java as a primary bioinformatics language. During this time, I'd wanted often to teach readseq to handle sequence documentation; the original ignored all but a few fields of information other than sequence data. In late 1990's my sequence analysis program SeqPup was in its Java incarnation and needed this, especially handling of feature annotations, locating genes, introns and such in a sequence.
After slowly updating readseq from a haphazard C program to an object oriented structure in Java, I pulled it back out of its parent source to become again a stand-alone program for format conversion. This release version 2, first available in 1999, continues support for the "classic" C version, in that it supports the same command-line options, but has extensions for sequence documentation, feature table and other additions, plus new sequence format conversions, and a lot of bug fixing. This java version is also more efficient, working faster than the compiled C classic version. It still isn't efficient enough to handle large sequences (genome sized or full GenBank/EMBL data release files).
In its current Java incarnation, interfacing Readseq with other languages is done mainly through command-line calls to the main program. If your programs are in Perl, you may want to use the bioperl.org collection with its SeqIO package.
The steps to use readseq are
Table Sequence formats recognized by format conversion program READSEQ
1. Abstract Syntax Notation (ASN.1)
2. DNA Strider
3. European Molecular Biology Laboratory (EMBL)
4. Fasta/Pearson
5. Fitch (for phylogenetic analysis)
6. GenBank
7. Genetics Computer Group (GCG)a
8. Intelligenetics/Stanford
9. Multiple sequence format (MSF)
10. National Biomedical Research Foundation (NBRF)
11. Olsen (in only)
12. Phylogenetic Analysis Using Parsimony (PAUP) NEXUS format
13. Phylogenetic Inference package (Phylip v3.3, v3.4)
14. Phylogenetic Inference package (Phylip v3.2)
15. Plain text/Stadena
16. Pretty format for publication (output only)
17. Protein Information Resource (PIR or CODATA)
18. Zuker for RNA analysis (in only)
a For conversion of single sequence files only. The other conversions can be performed on files with single or multiple sequences.
SEQIO, another sequence conversion program for a UNIX machine, is described at http://bioweb.pasteur.fr/docs/seqio/seqio.html and is available for download at http://www.cs.ucdavis.edu/_gusfield/seqio.html.
GCG PROGRAMS FOR CONVERSION OF SEQUENCE FORMATS:
The “from” programs convert sequence files from GCG format into the named format, and the “to” programs convert the alternative format into GCG format. Shown are the actual program names, no spaces included. There are no programs to convert to GenBank and EMBL formats.
FROMEMBL
FROMFASTA
FROMGENBANK
FROMIG
FROMPIR
FROMSTADEN
TOFASTA
TOIG
TOPIR
TOSTADEN
In addition, the GCG programs include the following sequence formatting programs: (1) GETSEQ, which converts a simple ASCII file being received from a remote PC to GCG format; (2) REFORMAT, which will format a GCG file that has been edited, and will also perform other functions; and (3) SPEW, which sends a GCG sequence file as an ASCII file to a remote PC.
EMBOSS:
EMBOSS is "The European Molecular Biology Open Software Suite". EMBOSS is a free Open Source software analysis package specially developed for the needs of the molecular biology (e.g. EMBnet) user community. The software automatically copes with data in a variety of formats and even allows transparent retrieval of sequence data from the web. Also, as extensive libraries are provided with the package, it is a platform to allow other scientists to develop and release software in true open source spirit. EMBOSS also integrates a range of currently available packages and tools for sequence analysis into a seamless whole. EMBOSS breaks the historical trend towards commercial software packages. Seqret is one of the program of EMBOSS package.
Seqret program reads and writes (returns) sequences. seqret can read a sequence or many sequences from databases, files, files of sequence names, the command-line or the output of other programs and then can write them to files, the screen or pass them to other programs. Because it can read in a sequence from a database and write it to a file, seqret is a program for extracting sequences from databases. Because it can write the sequence to the screen, seqret is a program for displaying sequences. seqret can read sequences in any of a wide range of standard sequence formats. You can specify the input and output formats being used. If you don't specify the input format, seqret will try a set of possible formats until it reads it in successfully. Because you can specify the output sequence format, seqret is a program to reformat a sequence. seqret can read in the reverse complement of a nucleic acid sequence. It therefore is a program for producing the reverse complement of a sequence. seqret can read in a sequence whose begin and end positions you have specified and write out that fragment. It is therefore a utility for doing simple extraction of a region of a sequence. seqret can change the case of the sequence being read in to upper or to lower case. It is therefore a simple sequence beautification utility. seqret can do any combination of the above functions.
To date, the following sequence formats are accepted as input.
|
Input Format |
Comments |
|
gcg |
GCG 9.x and 10.x format with the format and sequence type identified on the first line of the file |
|
gcg8 |
GCG 8.x format where anything up to the first line containing ".." is considered as heading, and the remainder is sequence data. This format is complicated by the header appearing to be in other formats such as EMBL, and by the possibility of reading a large amount of data in the wrong format before discovering that there is no ".." line because it is not GCG format after all. |
|
embl |
EMBL entry format, or at least a minimal subset of the fields. The Staden package and others use EMBL or similar formats for sequence data. |
|
swiss |
SWISSPROT entry format, or at least a minimal subset of the fields. |
|
fasta |
FASTA
format with an optional accession number after the sequence identifier, eg:
|
|
ncbi |
FASTA
format with optional accession number and database name in NCBI style
included as part of the sequence identifier. eg |
|
genbank |
GENBANK entry format, or at least a minimal subset of the fields. |
|
nbrf |
NBRF (PIR) format, as used in the PIR database sequence files. |
|
codata |
CODATA format. |
|
strider |
DNA strider format |
|
clustal |
ClustalW ALN (multiple alignment) format. |
|
phylip |
PHYLIP interleaved multiple alignment format. |
|
acedb |
ACeDB format |
|
msf |
Wisconsin Package GCG's MSF multiple sequence format. |
|
hennig86 |
Hennig86 format |
|
jackknifer |
Jackknifer format |
|
jackknifernon |
Jackknifernon format |
|
nexus |
Nexus/PAUP format |
|
nexusnon |
Nexusnon/PAUPnon format |
|
treecon |
Treecon format |
|
mega |
Mega format |
|
meganon |
Meganon format |
|
ig |
IntelliGenetics format. |
|
staden |
The experiment file format used by the "gap" program in the Staden package, where the sequence identifier is optional and the remainer is plain text. Some alternative nucleotide ambiguity codes are used and must be converted. |
|
unknown |
Plain text. This is the format with no format. The whole of the file is read in as a sequence. No attempt is made to parse the file contents in any way. Anything is acceptable in this format. |
|
raw |
Like unknown/text/plain format except that it accepts only alphanumeric and whitespace characters and rejects anything else. |
|
asis |
This is not
so much a sequence format as a quick way of entering a sequence on the
command line, but it is included here for completeness. Where a filename
would normally be given, in asis format there is the sequence itself.
An example would be: |
To date, the following sequence formats are available as output.
|
Output Format |
Single/ |
Comments |
|
gcg |
single |
Wisconsin Package GCG 9.x and 10.x format with the sequence type on the first line of the file. |
|
gcg8 |
single |
GCG 8.x format where anything up to the first line containing ".." is considered as heading, and the remainder is sequence data. |
|
embl |
multiple |
EMBL entry format with available fields filled in and others with no infomation omitted. The EMBOSS command line allows missing data such as accession numbers to be provided if they are not obtainable from the input sequence. |
|
swiss |
multiple |
SwisProt entry format with available fields filled in and others with no infomation omitted. The EMBOSS command line allows missing data such as accession numbers to be provided if they are not obtainable from the input sequence. |
|
fasta |
multiple |
Standard Pearson FASTA format, but with the accession number included after the identifier if available. |
|
pearson |
multiple |
Simple Pearson FASTA format, an alias for "fasta" format. |
|
ncbi |
multiple |
NCBI style FASTA format with the database name, entry name and accession number separated by pipe ("|") characters. |
|
nbrf |
multiple |
NBRF (PIR) format, as used in the PIR database sequence files. |
|
genbank |
multiple |
GENBANK entry format with available fields filled in and others with no infomation omitted. The EMBOSS command line allows missing data such as accession numbers to be provided if they are not obtainable from the input sequence. |
|
ig |
multiple |
Intelligenetics format, as used by the Intelligenetics package |
|
codata |
multiple |
CODATA format. |
|
strider |
multiple |
DNA strider format |
|
acedb |
multiple |
ACeDB format |
|
staden |
single |
The experiment file format used by the "gap" program in the Staden package. Some alternative nucleotide ambiguity codes are used and are converted. |
|
text |
single |
Plain sequence, no annotation or heading. |
|
fitch |
multiple |
Fitch format |
|
msf |
multiple |
Wisconsin Package GCG's MSF multiple sequence format. |
|
clustal |
multiple |
Clustal multiple sequence format. |
|
phylip |
multiple |
PHYLIP non-interleaved format. |
|
phylip3 |
multiple |
PHYLIP interleaved format. |
|
asn1 |
multiple |
A subset of ASN.1 containing entry name, accession number, description and sequence, similar to the current ASN.1 output of readseq |
|
hennig86 |
multiple |
Hennig86 format |
|
mega |
multiple |
Mega format |
|
meganon |
multiple |
Meganon format |
|
nexus |
multiple |
Nexus/PAUP format |
|
nexusnon |
multiple |
Nexusnon/PAUPnon format |
|
jackknifer |
multiple |
Jackknifer format |
|
jackknifernon |
multiple |
Jackknifernon format |
|
treecon |
multiple |
Treecon format |
|
debug |
multiple |
EMBOSS sequence object report for debugging showing all available fields. Not all fields will contain data - this depends very much on the input format used. |