BLOOD
COMPOSITION OF BLOOD
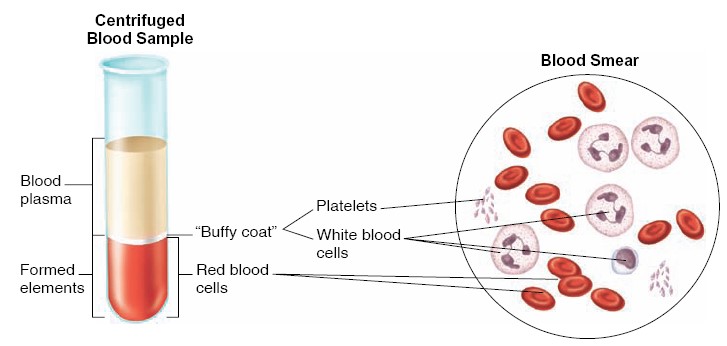
Blood consists of formed elements that are suspended and carried in a fluid
called plasma. The formed elements, erythrocytes, leukocytes, and platelets
function, respectively, in oxygen transport, immune defense, and blood clotting.
The total blood volume in the average-size adult is about 5 liters,
constituting about 8% of the total body weight. Blood leaving the heart is
referred to as arterial blood. Arterial blood, with the exception of that
going to the lungs, is bright red because of a high concentration of
oxyhemoglobin (the combination of oxygen and hemoglobin) in the red blood cells.
Venous blood is blood returning to the heart. Except for the venous blood
from the lungs, it contains less oxygen and
is therefore a darker red than the oxygen-rich arterial blood. Blood is composed
of a cellular portion, called formed elements, and a fluid portion,
called plasma. When a blood sample is centrifuged, the heavier formed
elements are packed into the bottom of the tube, leaving plasma at the top. The
formed elements constitute approximately 45% of the total blood volume, and the
plasma accounts for the remaining 55%. Red blood cells compose most of the
formed elements; the percentage of red blood cell volume to total blood volume
in a centrifuged blood sample (a measurement called the hematocrit) is
36% to 46% in women and 41% to 53% in men.
BLOOD COMPOSITION
Plasma
Plasma
is a straw-colored liquid consisting of water and dissolved solutes. The major
solute of the plasma in terms of its concentration is Na+. In
addition to Na+, plasma contains many other ions, as well as organic
molecules such as metabolites, hormones, enzymes, antibodies, and other
proteins.
Plasma Proteins
Plasma proteins
constitute 7% to 9% of the plasma. The three types of proteins are albumins,
globulins, and fibrinogen. Albumins account for most (60% to 80%) of the
plasma proteins and are the smallest in size. They are produced by the liver and
provide the osmotic pressure needed to draw water from the surrounding tissue
fluid into the capillaries. This action is needed to maintain blood volume and
pressure. Globulins are grouped into three subtypes: alpha globulins,
beta globulins, and gamma globulins. The alpha and beta globulins are
produced by the liver and function in transporting lipids and fat-soluble
vitamins. Gamma globulins are antibodies produced by lymphocytes (one of the
formed elements found in blood and lymphoid tissues) and function in immunity.
Fibrinogen, which accounts for only about 4% of the total plasma
proteins, is an important clotting factor produced by the liver. During the
process of clot formation, fibrinogen is converted into insoluble threads of
fibrin. Thus, the fluid from clotted blood, called serum, does not
contain fibrinogen but is otherwise identical to plasma.
|
Name |
Principal Function |
Binding Characteristics |
Serum or Plasma Concentration |
|
Albumin |
Binding and carrier protein; osmotic regulator |
Hormones, amino acids, steroids, vitamins, fatty acids |
4500–5000 mg/dL |
|
Orosomucoid |
Uncertain; may have a role in inflammation |
Trace; rises in inflammation |
|
|
α1-Antiprotease |
Trypsin and general protease inhibitor |
Proteases in serum and tissue secretions |
1.3–1.4 mg/dL |
|
α-Fetoprotein |
Osmotic regulation; binding and carrier proteina |
Hormones, amino acids |
Found normally in fetal blood |
|
α2-Macroglobulin |
Inhibitor of serum endoproteases |
Proteases |
150–420 mg/dL |
|
Antithrombin-III |
Protease inhibitor of intrinsic coagulation system |
1:1 binding to proteases |
17–30 mg/dL |
|
Ceruloplasmin |
Transport of copper |
Six atoms copper/molecule |
15–60 mg/dL |
|
C-reactive protein |
Uncertain; has role in tissue inflammation |
Complement C1q |
<1 mg/dL; rises in inflammation |
|
Fibrinogen |
Precursor to fibrin in hemostasis |
200–450 mg/dL |
|
|
Haptoglobin |
Binding, transport of cell-free hemoglobin |
Hemoglobin 1:1 binding |
40–180 mg/dL |
|
Hemopexin |
Binds to porphyrins, particularly heme for heme recycling |
1:1 with heme |
50–100 mg/dL |
|
Transferrin |
Transport of iron |
Two atoms iron/molecule |
3.0–6.5 mg/dL |
|
Apolipoprotein B |
Assembly of lipoprotein particles |
Lipid carrier |
|
|
Angiotensinogen |
Precursor to pressor peptide angiotensin II |
||
|
Proteins, coagulation factors II, VII, IX, X |
Blood clotting |
20 mg/dL |
|
|
Antithrombin C, protein C |
Inhibition of blood clotting |
||
|
Insulinlike growth factor I |
Mediator of anabolic effects of growth hormone |
IGF-I receptor |
|
|
Steroid hormone-binding globulin |
Carrier protein for steroids in bloodstream |
Steroid hormones |
3.3 mg/dL |
|
Thyroxine-binding globulin |
Carrier protein for thyroid hormone in bloodstream |
Thyroid hormones |
1.5 mg/dL |
|
Transthyretin (thyroid-binding prealbumin) |
Carrier protein for thyroid hormone in bloodstream |
Thyroid hormones |
25 mg/dL |
|
a The function of α-fetoprotein is uncertain, but because of its
structural homology to albumin it is often assigned these functions. |
|||
Plasma Volume
A number of regulatory mechanisms in the body maintain homeostasis of the plasma
volume. If the body should lose water, the remaining plasma becomes excessively
concentrated—its osmolality increases. This is
detected by osmoreceptors in the hypothalamus, resulting in a sensation of
thirst and the release of antidiuretic hormone (ADH) from the posterior
pituitary. This hormone promotes water retention by the kidneys, which—together
with increased intake of fluids—helps compensate for the dehydration and lowered
blood volume. This regulatory mechanism, together with others that influence
plasma volume, are very important in maintaining blood pressure.
The Formed Elements of Blood
The formed elements of blood include two types of blood cells:
erythrocytes, or red blood cells, and leukocytes, or white
blood cells. Erythrocytes are by far the more numerous of the two. A cubic
millimeter of blood normally contains 5.1 million to 5.8 million erythrocytes in
males and 4.3 million to 5.2 million erythrocytes in females. By contrast, the
same volume of blood contains only 5,000 to 9,000 leukocytes.
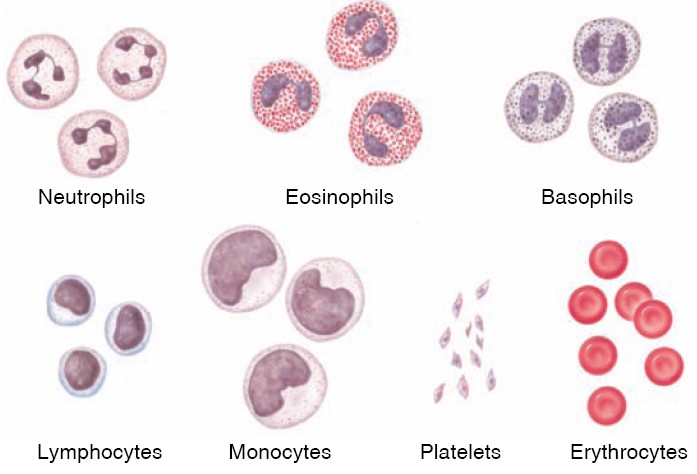
Leukocytes
Leukocytes
differ from erythrocytes in several respects. Leukocytes contain nuclei and
mitochondria and can move in an amoeboid fashion. Because of their amoeboid
ability, leukocytes can squeeze through pores in capillary walls and move to a
site of infection, whereas erythrocytes usually remain confined within blood
vessels. The movement of leukocytes through capillary walls is referred to as
diapedesis or extravasation. White blood cells are almost invisible
under the microscope unless they are stained; therefore, they are classified
according to their staining properties. Those leukocytes that have granules in
their cytoplasm are called granular leukocytes; those without clearly
visible granules are called agranular (or nongranular)
leukocytes. The stain used to identify white blood cells is usually a
mixture of a pink-to-red stain called eosin and a blue-topurple stain
(methylene blue), which is called a “basic stain.” Granular leukocytes with
pink-staining granules are therefore called eosinophils, and those with
blue-staining granules are called basophils. Those with granules that
have little affinity for either stain are neutrophils. Neutrophils are
the most abundant type of leukocyte, accounting for 50% to 70% of the leukocytes
in the blood. Immature neutrophils have sausage-shaped nuclei and are called
band cells. As the band cells mature, their nuclei become lobulated, with
two to five lobes connected by thin strands. At this stage, the neutrophils are
also known as polymorphonuclear leukocytes (PMNs). There are two types of
agranular leukocytes: lymphocytes and monocytes. Lymphocytes are usually
the second most numerous type of leukocyte; they are small cells with round
nuclei and little cytoplasm. Monocytes, by contrast, are the largest of
the leukocytes and generally have kidney- or horseshoe-shaped nuclei. In
addition to these two cell types, there are smaller numbers of plasma cells,
which are derived from lymphocytes. Plasma cells produce and secrete large
amounts of antibodies.
Blood cells:
Lymph
Lymph is tissue fluid that enters the lymphatic vessels. It drains into the
venous blood via the thoracic and right lymphatic ducts. It contains clotting
factors and clots on standing in vitro. In most locations, it also contains
proteins that have traversed capillary walls and can then return to the blood
via the lymph. Nevertheless, its protein content is generally lower than that of
plasma, which contains about 7 g/dL, but lymph protein content varies with the
region from which the lymph drains. Water-insoluble fats are absorbed from the
intestine into the lymphatics, and the lymph in the thoracic duct after a meal
is milky because of its high fat content. Lymphocytes also enter the circulation
principally through the lymphatics, and there are appreciable numbers of
lymphocytes in thoracic duct lymph.
Erythrocytes
The red blood cells (erythrocytes) carry hemoglobin in the circulation.
They are biconcave disks
that are manufactured in the bone marrow. In mammals, they lose their nuclei
before entering the circulation. In humans, they survive in the circulation for
an average of 120 days. The average normal red blood cell count is 5.4
million/μL in men and 4.8 million/μL in women. The number of red cells is also
conveniently expressed as the hematocrit, or the percentage of the blood,
by volume that is occupied by erythrocytes. Each human red blood cell is about
7.5 μm in diameter and 2 μm thick, and each contains approximately 29 pg of
hemoglobin. There are thus about 3 × 1013
red blood cells and about 900 g of hemoglobin in the circulating
blood of an adult man.
Haematopoiesis
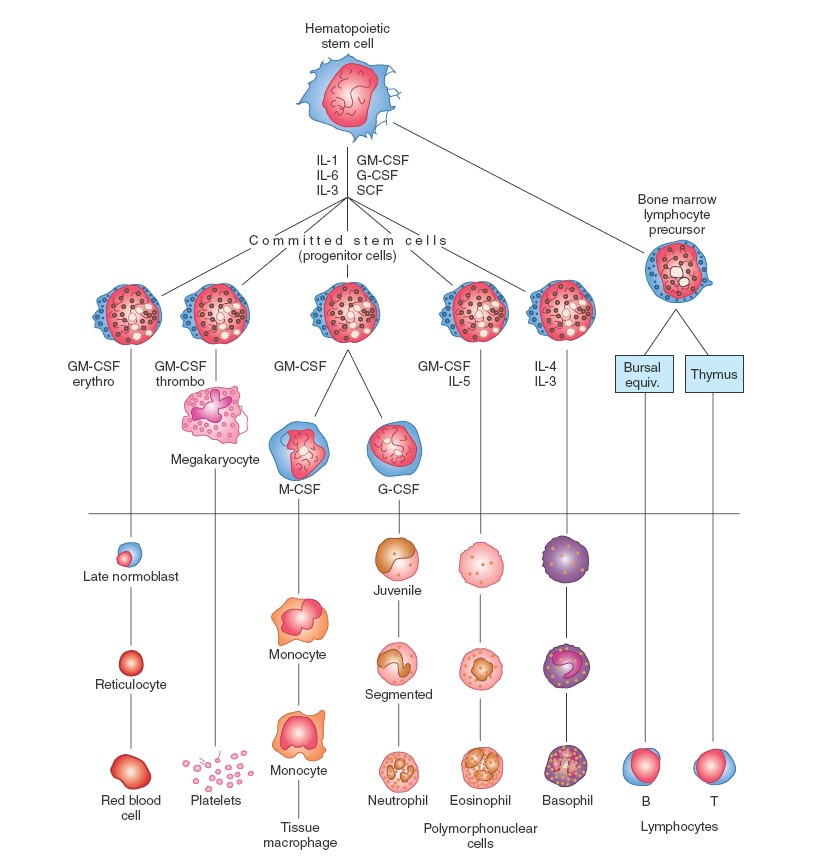
Platelets
Platelets are small, granulated bodies that aggregate at sites of vascular
injury. They lack nuclei and are 2–4 μm in diameter. There are about 300,000/μL
of circulating blood, and they normally have a half-life of about 4 days. The
megakaryocytes, giant cells in the bone marrow, form platelets by pinching
off bits of cytoplasm and extruding them into the circulation. Between 60% and
75% of the platelets that have been extruded from the bone marrow are in the
circulating blood, and the remainder are mostly in the spleen. Splenectomy
causes an increase in the platelet count (thrombocytosis).
Composition of Lymph
|
Source of Lymph |
Protein Content (g/dL) |
|
Choroid plexus |
0 |
|
Ciliary body |
0 |
|
Skeletal muscle |
2 |
|
Skin |
2 |
|
Lung |
4 |
|
Gastrointestinal tract |
4.1 |
|
Heart |
4.4 |
|
Liver |
6.2 |
FORMED ELEMENTS OF BLOOD
|
Component |
Description |
Number Present
|
Function |
|
Erythrocyte (red blood cell) |
Biconcave disc without nucleus; contains hemoglobin; survives 100 to 120
days |
4,000,000 to 6,000,000 / mm3 |
Transports oxygen and carbon dioxide |
|
Leukocytes (white blood cells) |
|
5,000 to 10,000 / mm3 |
Aid in defense against infections by microorganisms |
|
Granulocytes |
About twice the size of red blood cells; cytoplasmic granules present;
survive 12 hours to 3 days |
|
|
|
1. Neutrophil |
Nucleus with 2 to 5 lobes; cytoplasmic granules stain slightly pink |
54% to 62% of white cells present |
Phagocytic |
|
2. Eosinophil |
Nucleus bilobed; cytoplasmic granules stain red in eosin stain |
1% to 3% of white cells present |
Helps to detoxify foreign substances; secretes enzymes that dissolve
clots; fights parasitic infections |
|
3. Basophil |
Nucleus lobed; cytoplasmic granules stain blue in hematoxylin stain |
Less than 1% of white cells present |
Releases anticoagulant heparin |
|
Agranulocytes |
Cytoplasmic granules not visible; survive 100 to 300 days (some much
longer) |
|
|
|
1. Monocyte |
2 to 3 times larger than red blood cell; nuclear shape varies from round
to lobed |
3% to 9% of white cells present |
Phagocytic |
|
2. Lymphocyte |
Only slightly larger than red blood cell; nucleus nearly fits cell |
25% to 33% of white cells present |
Provides specific immune response (including antibodies) |
|
Platelet (thrombocyte) |
Cytoplasmic fragment; survives 5 to 9 days |
130,000 to 400,000 / mm3 |
Enables clotting; releases serotonin, which causes vasoconstriction |
BLOOD GROUPS
There are certain molecules on the surfaces of all cells in the body that can be
recognized as foreign by the immune system of another individual. These
molecules are known as antigens. As part of the immune response,
particular lymphocytes secrete a class of proteins called antibodies that
bond in a specific fashion with antigens. The specificity of antibodies for
antigens is analogous to the specificity of enzymes for their substrates, and of
receptor proteins for neurotransmitters and hormones.
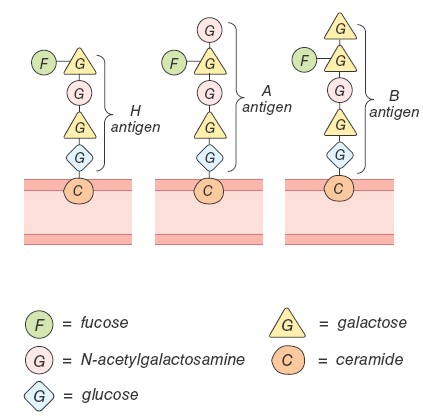
ABO System
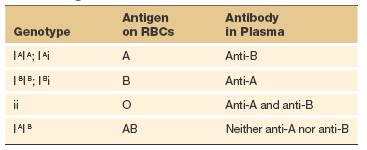
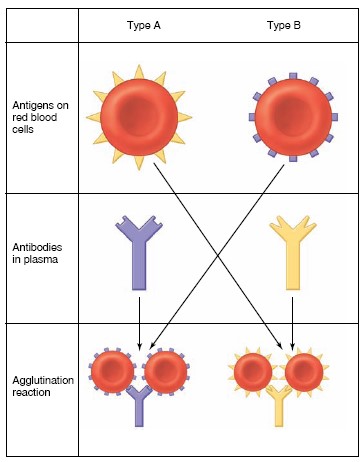
The distinguishing antigens on other cells are far more varied than the antigens
on red blood cells. Red blood cell antigens, however, are of extreme clinical
importance because their types must be matched between donors and recipients for
blood transfusions. There are several groups of red blood cell antigens, but the
major group is known as the ABO system. In terms of the antigens present
on the red blood cell surface, a person may be type A (with only A
antigens), type B (with only B antigens), type AB (with both A and
B antigens), or type O (with neither A nor B antigens). Each person’s
blood type-A, B, or O-denotes the antigens present on the red blood cell
surface, which are the products of the genes (located on chromosome number 9)
that code for these antigens.
Each person inherits two genes (one from each parent) that control the
production of the ABO antigens. The genes for A or B antigens are dominant to
the gene for O. The O gene is recessive, simply because it doesn’t code for
either the A or the B red blood cell antigens. The genes for A and B are often
shown as I A and I B and the
recessive gene for O is shown as the lowercase i. A person who is type A,
therefore, may have inherited the A gene from each parent (may have the genotype
I A I A ), or the A gene from one parent and the O gene from the other parent
(and thus have the genotype I A i). Likewise, a person who is type B may have
the genotype I B I B or I B i. It follows that a type O person inherited the O
gene from each parent (has the genotype ii), whereas a type AB person inherited
the A gene from one parent and the B gene from the other (there is no
dominant-recessive relationship between A and B). The immune system exhibits
tolerance to its own red blood cell antigens. People who are type A, for
example, do not produce anti-A antibodies. Surprisingly, however, they do make
antibodies against the B antigen and, conversely, people with blood type B make
antibodies against the A antigen.
This is believed to result from the fact that antibodies made in response to
some common bacteria cross-react with the A or B antigens. People who are type
A, therefore, acquire antibodies that can react with B antigens by exposure to
these bacteria, but they do not develop anti bodies that can react with A
antigens because tolerance mechanisms prevent this. People who are type AB
develop tolerance to both of these antigens, and thus do not produce either
anti-A or anti-B antibodies. Those who are type O, by contrast, do not develop
tolerance to either antigen; therefore, they have both anti-A and anti-B
antibodies in their plasma.
Transfusion Reactions
Before transfusions are performed, a major crossmatch is made by mixing
serum from the recipient with blood cells from the donor. If the types do not
match—if the donor is type A, for example, and the recipient is type B—the
recipient’s antibodies attach to the donor’s red blood cells and form bridges
that cause the cells to clump together, or agglutinate. Because of this
agglutination reaction, the A and B antigens are sometimes called
agglutinogens, and the antibodies against them are called agglutinins.
Transfusion errors that result in such agglutination can lead to blockage of
small blood vessels and cause hemolysis (rupture of red blood cells), which may
damage the kidneys and other organs. In emergencies, type O blood has been given
to people who are type A, B, AB, or O. Because type O red blood cells lack A and
B antigens, the recipient’s antibodies cannot cause agglutination of the donor
red blood cells. Type O is, therefore, a universal donor —but only as
long as the volume of plasma donated is small, since plasma from a type O person
would agglutinate type A, type B, and type AB red blood cells. Likewise, type AB
people are universal recipients because they lack anti-A and anti-B
antibodies, and thus cannot agglutinate donor red blood cells. (Donor plasma
could agglutinate recipient red blood cells if the transfusion volume were too
large.) Because of the dangers involved, use of the universal donor and
recipient concept is strongly discouraged in practice.
Rh Factor
Another group of antigens found on the red blood cells of most people is the
Rh factor (named for the rhesus monkey, in which these antigens were first
discovered). There are a number of different antigens in this group, but one
stands out because of its medical significance. This Rh antigen is termed D, and
is often indicated as Rho(D). If this Rh antigen
is present on a person’s red blood cells, the person is Rh positive; if
it is absent, the person is Rh negative. The Rh-positive condition is by
far the more common (with a frequence of 85% in the Caucasian population, for
example). The Rh factor is of particular significance when Rh- negative mothers
give birth to Rh-positive babies. The fetal and maternal blood are normally kept
separate across the placenta, and so the Rh-negative mother is not usually
exposed to the Rh antigen of the fetus during the pregnancy. At the time of
birth, however, a variable degree of exposure may occur, and the mother’s immune
system may become sensitized and produce antibodies against the Rh antigen. This
does not always occur, however, because the exposure may be minimal and because
Rh-negative women vary in their sensitivity to the Rh factor. If the woman does
produce antibodies against the Rh factor, these antibodies could cross the
placenta in subsequent pregnancies and cause hemolysis of the Rh-positive red
blood cells of the fetus. Therefore, the baby could be born anemic with a
condition called erythroblastosis fetalis, or hemolytic disease of the
newborn. Erythroblastosis fetalis can be prevented by injecting the
Rh-negative mother with an antibody preparation against the Rh factor (a trade
name for this preparation is RhoGAM—the GAM is short for gamma globulin, the
class of plasma proteins in which antibodies are found) within 72 hours after
the birth of each Rh-positive baby. This is a type of passive immunization in
which the injected antibodies inactivate the Rh antigens and thus prevent the
mother from becoming actively immunized to them. Some physicians now give RhoGAM
throughout the Rh-positive pregnancy of any Rh-negative woman.
AGGLUTINATIONS REACTIONS
BLOOD TYPING
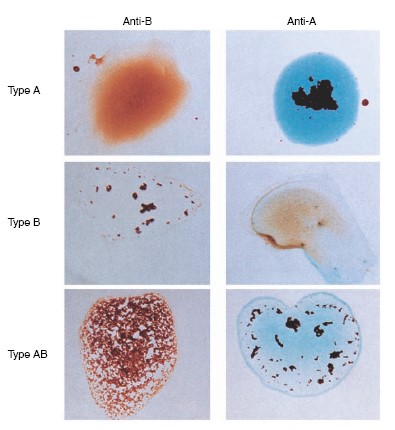
BLOOD CLOTTING
More than 50 important substances that cause or affect blood coagulation have
been found in the blood and in the tissues—some that promote coagulation, called
procoagulants, and others that inhibit coagulation, called
anticoagulants. Whether blood will coagulate depends on the balance between
these two groups of substances. In the blood stream, the anticoagulants normally
predominate, so the blood does not coagulate while it is circulating in the
blood vessels. However, when a vessel is ruptured, procoagulants from the area
of tissue damage become “activated” and override the anticoagulants, and then a
clot does develop.
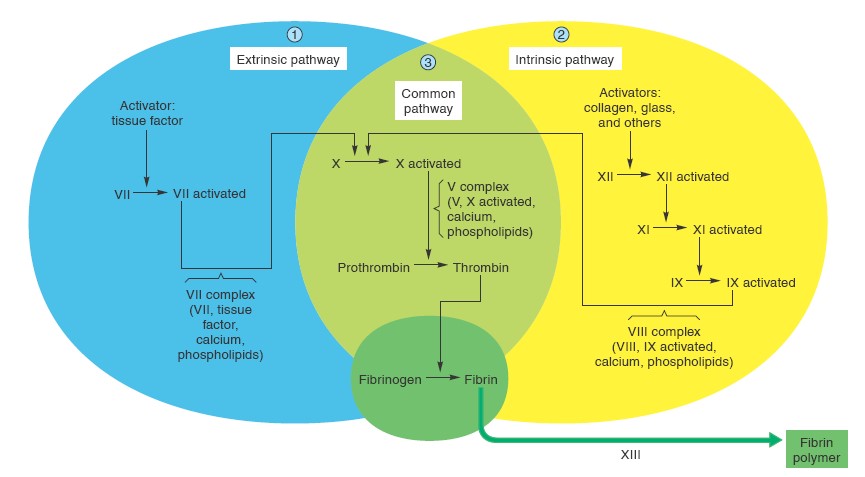
Clotting takes place in three essential steps:
1. In response to rupture of the vessel or damage to the blood itself, a complex
cascade of chemical reactions occurs in the blood involving more than a dozen
blood coagulation factors. The net result is formation of a complex of activated
substances collectively called prothrombin activator.
2. The prothrombin activator catalyzes conversion of prothrombin into
thrombin.
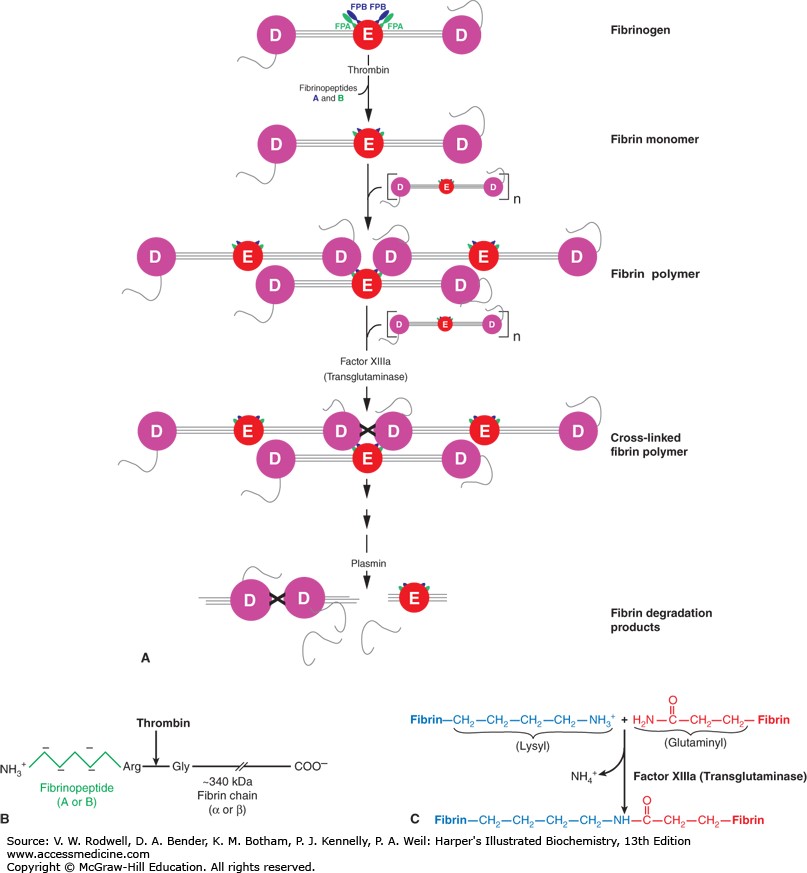
3. The thrombin acts as an enzyme to convert fibrinogen into fibrin
fibers that enmesh platelets, blood cells, and plasma to form the clot.
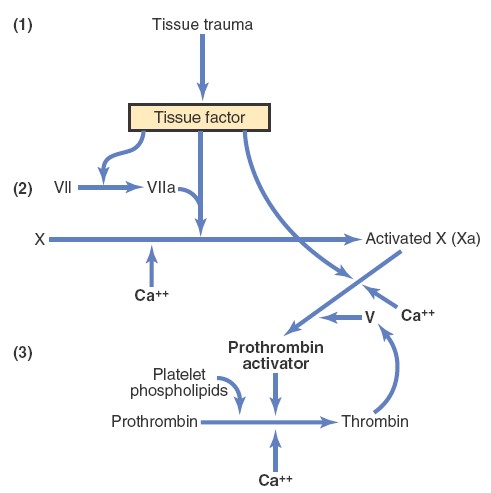
INITIATION OF COAGULATION - FORMATION OF PROTHROMBIN ACTIVATOR
These mechanisms are set into play by (1) trauma to the vascular wall and
adjacent tissues, (2) trauma to the blood, or (3) contact of the blood with
damaged endothelial cells or with collagen and other tissue elements outside the
blood vessel. In each instance, this leads to the formation of prothrombin
activator, which then causes prothrombin conversion to thrombin and all the
subsequent clotting steps. Prothrombin activator is generally considered to be
formed in two ways, although, in reality, the two ways interact constantly with
each other: (1) by the extrinsic pathway that begins with trauma to the
vascular wall and surrounding tissues and (2) by the intrinsic pathway
that begins in the blood. In both the extrinsic and the intrinsic pathways, a
series of different plasma proteins called blood-clotting factors plays a
major role. Most of these proteins are inactive forms of proteolytic
enzymes. When converted to the active forms, their enzymatic actions cause the
successive, cascading reactions of the clotting process. Most of the clotting
factors, which are designated by Roman numerals. To indicate the activated form
of the factor, a small letter “a” is added after the Roman numeral, such as
Factor VIIIa to indicate the activated state of Factor VIII.
Extrinsic Pathway for Initiating Clotting
The extrinsic pathway for initiating the formation of prothrombin activator
begins with a traumatized vascular wall or traumatized extravascular tissues
that come in contact with the blood. This condition leads to the following
steps:
1. Release of tissue factor. Traumatized tissue releases a complex of
several factors called tissue factor or tissue thromboplastin.
This factor is composed especially of phospholipids from the membranes of
the tissue plus a lipoprotein complex that functions mainly as a
proteolytic enzyme.
2. Activation of Factor X—role of Factor VII and tissue factor. The
lipoprotein complex of tissue factor further complexes with blood coagulation
Factor VII and, in the presence of calcium ions, acts enzymatically on Factor X
to form activated Factor X (Xa).
3. Effect of Xa to form prothrombin activator—role of Factor V. The
activated Factor X combines immediately with tissue phospholipids that are part
of tissue factors or with additional phospholipids released from platelets, as
well as with Factor V, to form the complex called prothrombin activator.
Within a few seconds, in the presence of Ca++,
prothrombin is split to form thrombin, and the clotting process proceeds as
already explained. At first, the Factor V in the prothrombin activator complex
is inactive, but once clotting begins and thrombin begins to form, the
proteolytic action of thrombin activates Factor V. This activation then becomes
an additional strong accelerator of prothrombin activation. Thus, in the final
prothrombin activator complex, activated Factor X is the actual protease that
causes splitting of prothrombin to form thrombin; activated Factor V greatly
accelerates this protease activity, and platelet phospholipids act as a vehicle
that further accelerates the process. Note especially the positive feedback
effect of thrombin, acting through Factor V, to accelerate the entire
process once it begins.
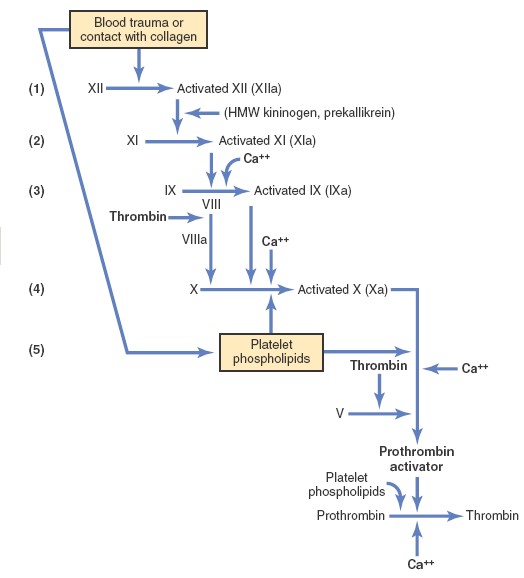
Intrinsic Pathway for Initiating Clotting
The second mechanism for initiating formation of prothrombin activator, and
therefore for initiating clotting, begins with trauma to the blood or
exposure of the blood to collagen from a traumatized blood vessel wall. Then
the process continues through the series of cascading reactions.
1. Blood trauma causes (1) activation of Factor XII and (2) release of
platelet phospholipids. Trauma to the blood or exposure of the blood to
vascular wall collagen alters two important clotting factors in the blood:
Factor XII and the platelets. When Factor XII is disturbed, such as by coming
into contact with collagen or with a wettable surface such as glass, it takes on
a new molecular configuration that converts it into a proteolytic enzyme called
“activated Factor XII.” Simultaneously, the blood trauma also damages the
platelets because of adherence to
either collagen or a wettable surface (or by damage in other ways), and this
releases platelet phospholipids that contain the lipoprotein called platelet
factor 3, which also plays a role in subsequent clotting reactions.
2. Activation of Factor XI. The activated Factor XII acts enzymatically
on Factor XI to activate this factor as well, which is the second step in the
intrinsic pathway. This reaction also requires highmolecular-weight kininogen
and is accelerated by prekallikrein.
3. Activation of Factor IX by activated Factor XI. The activated Factor
XI then acts enzymatically on Factor IX to activate this factor as well.
4. Activation of Factor X—role of Factor VIII. The activated Factor IX,
acting in concert with activated Factor VIII and with the platelet phospholipids
and Factor III from the traumatized platelets, activates Factor X. It is clear
that when either Factor VIII or platelets are in short supply, this step is
deficient. Factor VIII is the factor that is missing in a person who has classic
hemophilia, for which reason it is called antihemophilic factor.
Platelets are the clotting factor that is lacking in the bleeding disease called
thrombocytopenia.
5. Action of activated Factor X to form prothrombin activator—role of Factor
V. This step in the intrinsic pathway is the same as the last step in the
extrinsic pathway. That is, activated Factor X combines with Factor V and
platelet or tissue phospholipids to form the complex called prothrombin
activator. The prothrombin activator in turn initiates within seconds the
cleavage of prothrombin to form thrombin, thereby setting into motion the final
clotting process, as described earlier.
Interaction between the Extrinsic and Intrinsic Pathways
It is clear from the schemas of the intrinsic and extrinsic systems that after
blood vessels rupture, clotting occurs by both pathways simultaneously. Tissue
factor initiates the extrinsic pathway, whereas contact of Factor XII and
platelets with collagen in the vascular wall initiates
the intrinsic pathway.
An especially important difference between the extrinsic and intrinsic pathways
is that the extrinsic pathway can be explosive; once initiated, its speed
of completion to the final clot is limited only by the amount of tissue factor
released from the traumatized tissues and by the quantities of Factors X, VII,
and V in the blood. With severe tissue trauma, clotting can occur in as little
as 15 seconds. The intrinsic pathway is much slower to proceed, usually
requiring 1 to 6 minutes to cause clotting.
Clotting factors
|
Clotting Factor
|
Synonyms
|
|
Fibrinogen |
Factor I |
|
Prothrombin |
Factor II |
|
Tissue factor |
Factor III; tissue thromboplastin |
|
Calcium |
Factor IV |
|
Factor V |
Proaccelerin; labile factor; Ac-globulin (Ac-G) |
|
Factor VII |
Serum prothrombin conversion accelerator (SPCA); proconvertin; stable
factor |
|
Factor VIII |
Antihemophilic factor (AHF); antihemophilic globulin (AHG);
antihemophilic factor A |
|
Factor IX |
Plasma thromboplastin component (PTC); Christmas factor; antihemophilic
factor B. This factor named with Stephen Christmas who was first
identified with Christmas disease in 1952. |
|
Factor X |
Stuart factor; Stuart-Prower factor |
|
Factor XI |
Plasma thromboplastin antecedent (PTA); antihemophilic factor C |
|
Factor XII |
Hageman factor
|
|
Factor XIII |
Fibrin-stabilizing factor |
|
Prekallikrein |
Fletcher factor |
|
High-molecular-weight kininogen |
Fitzgerald factor; HMWK (high-molecular-weight kininogen) |
|
Platelets |
Closed wall formation due to its aggregation |
|
Von-Willebrand factor |
It binds to factor VIII and prevent its degradation. It also binds to
collagen of vascular injury site and promotes platelets attachment and
aggregation. |
EXTRINSIC PATHWAY
INTRINSIC PATHWAY
DISEASES DUE TO BLOOD CLOTTING
|
Deficiency of Factor: |
Clinical Syndrome |
Cause |
|
I |
Afibrinogenemia |
Depletion during pregnancy with premature separation of placenta; also
congenital (rare) |
|
II |
Hypoprothrombinemia (hemorrhagic tendency in liver disease) |
Decreased hepatic synthesis, usually secondary to vitamin K deficiency |
|
V |
Parahemophilia |
Congenital |
|
VII |
Hypoconvertinemia |
Congenital |
|
VIII |
Hemophilia A (classic hemophilia) |
Congenital defect due to various abnormalities of the gene on X
chromosome that codes for factor VIII; disease is therefore inherited as
a sex-linked characteristic |
|
IX |
Hemophilia B (Christmas disease) |
Congenital |
|
X |
Stuart-Prower factor deficiency |
Congenital |
|
XI |
PTA deficiency |
Congenital |
|
XII |
Hageman trait |
Congenital |
BLEEDING TIME
When a sharp-pointed knife is used to pierce the tip of the finger or lobe of
the ear, bleeding ordinarily lasts for 1 to 6 minutes. The time depends largely
on the depth of the wound and the degree of hyperemia in the finger or ear lobe
at the time of the test. Lack of any one of several of the clotting factors can
prolong the bleeding time, but it is especially prolonged by lack of platelets.
CLOTTING TIME
Many methods have been devised for determining blood clotting times. The one
most widely used is to collect blood in a chemically clean glass test tube and
then to tip the tube back and forth about every 30 seconds until the blood has
clotted. By this method, the normal clotting time is 6 to 10 minutes. Procedures
using multiple test tubes have also been devised for determining clotting time
more accurately. Unfortunately, the clotting time varies widely, depending on
the method used for measuring it, so it is no longer used in many clinics.
Instead, measurements of the clotting factors themselves are made, using
sophisticated chemical procedures.
Prothrombin time gives an indication of the concentration of prothrombin in the
blood.
CIRCULATORY SYSTEM
BASIC ANATOMY OF HEART
The heart is a muscular organ enclosed in a protective fibrous sac, the
pericardium, and located in the chest. A fibrous layer is also closely
affixed to the heart and is called the epicardium. The extremely narrow
space between the pericardium and the epicardium is filled with a watery fluid
that serves as a lubricant as the heart moves within the sac. The wall of the
heart, the myocardium, is composed primarily of cardiac muscle cells. The
inner surface of the cardiac chambers, as well as the inner wall of all blood
vessels, is lined by a thin layer of cells known as endothelial cells, or
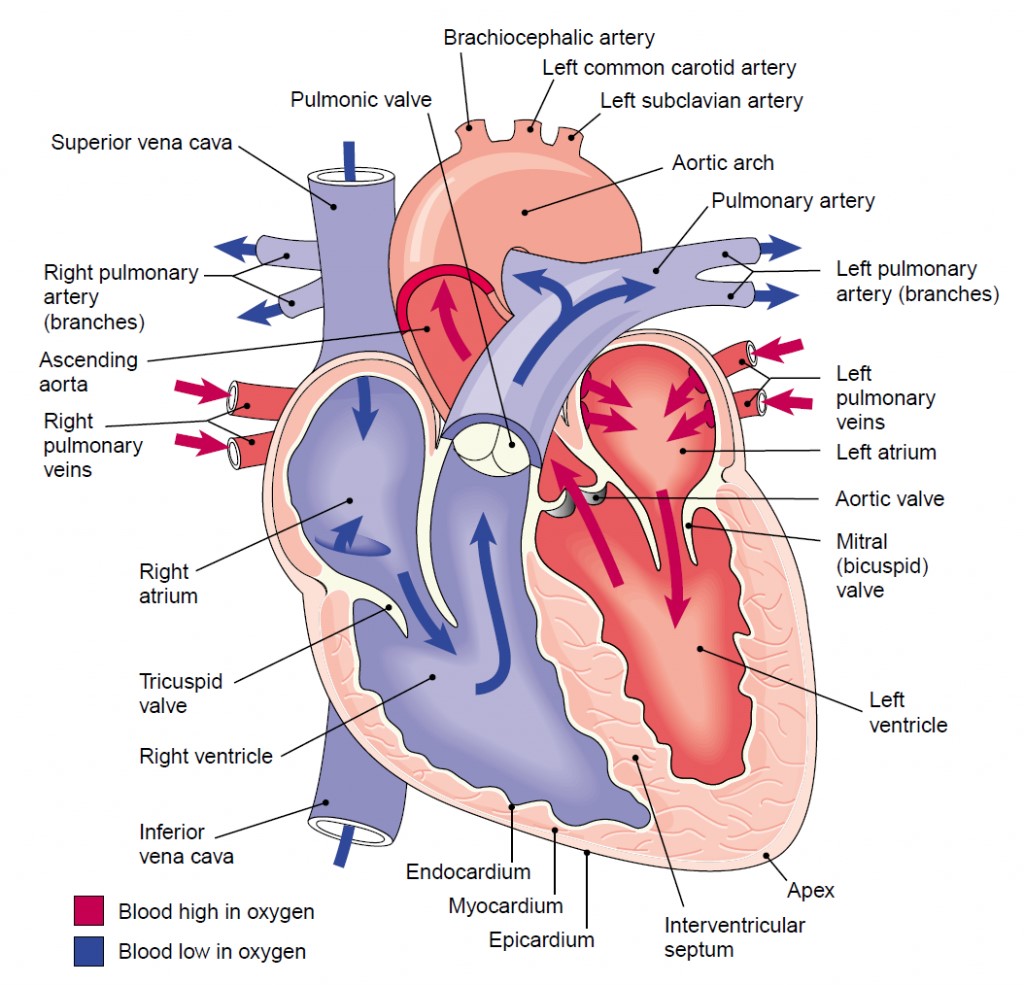
endothelium. As noted earlier, the human heart is divided into right and
left halves, each consisting of an atrium and a ventricle. The two ventricles
are separated by a muscular wall, the interventricular septum. Located
between the atrium and ventricle in each half of the heart are the one-way
atrioventricular (AV) valves, which permit blood to flow from
atrium to ventricle but not backward from ventricle to atrium. The right AV
valve is called the tricuspid valve because it has three fibrous flaps,
or cusps. The left AV valve has two flaps and is therefore called the
bicuspid valve. Its resemblance to a bishop’s headgear (a “mitre”) has
earned the left AV valve another commonly used name, mitral valve. The
opening and closing of the AV valves are passive processes resulting from
pressure differences across the valves. When the blood pressure in an atrium is
greater than in the corresponding ventricle, the valve is pushed open and blood
flows from atrium to ventricle. In contrast, when a contracting ventricle
achieves an internal pressure greater than that in its connected atrium, the AV
valve between them is forced closed. Therefore, blood does not normally move
back into the atria but is forced into the pulmonary trunk from the right
ventricle and into the aorta from the left ventricle.
To prevent the AV valves from being pushed up and opening backward into the
atria when the ventricles are contracting (a condition called prolapse),
the valves are fastened to muscular projections (papillary muscles) of
the ventricular walls by fibrous strands (chordae tendineae). The
papillary muscles do not open or close the valves. They act only to limit the
valves’ movements and prevent the backward flow of blood. Injury and disease of
these tendons or muscles can lead to prolapse. The openings of the right
ventricle into the pulmonary trunk and of the left ventricle into the aorta also
contain valves, the pulmonary and aortic valves, respectively.
These valves are also referred to as the semilunar valves, due to the half-moon
shape of the cusps. These valves allow blood to flow into the arteries during
ventricular contraction but prevent blood from moving in the opposite direction
during ventricular relaxation. Like the AV valves, they act in a passive manner.
Whether they are open or closed depends upon the pressure differences across
them. Another important point concerning the heart valves is that, when open,
they offer very little resistance to flow. Consequently, very small pressure
differences across them suffice to produce large flows. In disease states,
however, a valve may become narrowed or not open fully so that it offers a high
resistance to flow even when open. In such a state, the contracting cardiac
chamber must produce an unusually high pressure to cause flow across the valve.
There are no valves at the entrances of the superior and inferior venae cavae
(singular, vena cava) into the right atrium, and of the pulmonary veins
into the left atrium. However, atrial contraction pumps very little blood back
into the veins because atrial contraction constricts their sites of entry into
the atria, greatly increasing the resistance to backflow.
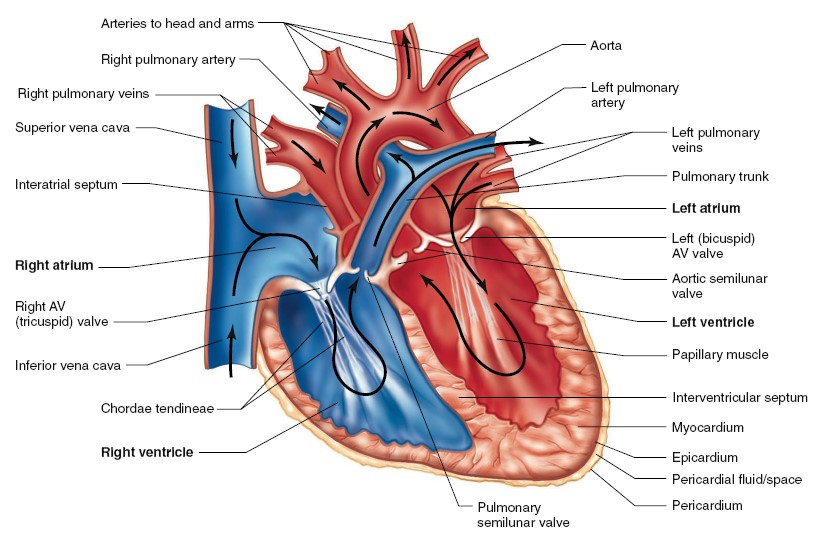
Pulmonary and Systemic Circulations
Blood whose oxygen content has become partially depleted and whose carbon
dioxide content has increased as a result of tissue metabolism returns to the
right atrium. This blood then enters the right ventricle, which pumps it into
the pulmonary trunk and pulmonary arteries. The pulmonary arteries
branch to transport blood to the lungs, where gas exchange occurs between the
lung capillaries and the air sacs (alveoli) of the lungs. Oxygen diffuses from
the air to the capillary blood, while carbon dioxide diffuses in the opposite
direction.
The blood that returns to the left atrium by way of the pulmonary veins
is therefore enriched in oxygen and partially depleted of carbon dioxide. The
path of blood from the heart (right ventricle), through the lungs, and back to
the heart (left atrium) completes one circuit: the pulmonary circulation.
Oxygen-rich blood in the left atrium enters the left ventricle and is pumped
into a very large, elastic artery-the aorta. The aorta ascends for a
short distance, makes a U-turn, and then descends through the thoracic (chest)
and abdominal cavities. Arterial branches from the aorta supply oxygen-rich
blood to all of the organ systems and are thus part of the systemic
circulation.
As a result of cellular respiration, the oxygen concentration is lower and the
carbon dioxide concentration is higher in the tissues than in the capillary
blood. Blood that drains from the tissues into the systemic veins is thus
partially depleted of oxygen and increased in carbon dioxide content. These
veins ultimately empty into two large veins—the superior and inferior
venae cavae —that return the oxygen-poor blood to the right atrium. This
completes the systemic circulation: from the heart (left ventricle), through the
organ systems, and back to the heart (right atrium). The numerous small muscular
arteries and arterioles of the systemic circulation present greater resistance
to blood flow than that in the pulmonary circulation. Despite the differences in
resistance, the rate of blood flow through the systemic circulation must be
matched to the flow rate of the pulmonary circulation. Because the amount of
work performed by the left ventricle is greater (by a factor of 5 to 7) than
that performed by the right ventricle, it is not surprising that the muscular
wall of the left ventricle is thicker (8 to 10 mm) than that of the right
ventricle (2 to 3 mm).
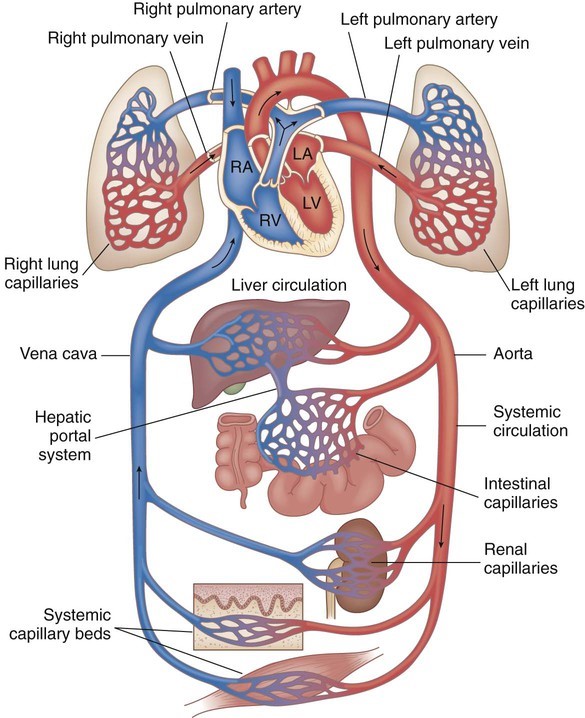
MECHANICAL EVENTS OF THE CARDIAC CYCLE
EVENTS IN LATE DIASTOLE
Late in diastole, the mitral (bicuspid) and tricuspid valves between the atria
and ventricles (atrioventricular [AV] valves) are open and the aortic and
pulmonary valves are closed. Blood flows into the heart throughout diastole,
filling the atria and ventricles. The rate of filling declines as the
ventricles become distended and, especially when the heart rate is low, the
cusps of the AV valves drift toward the closed position. The pressure in the
ventricles remains low. About 70% of the ventricular filling occurs passively
during diastole.
ATRIAL SYSTOLE
Contraction of the atria propels some additional blood into the ventricles.
Contraction of the atrial muscle narrows the orifices of the superior and
inferior vena cava and pulmonary veins, and the inertia of the blood moving
toward the heart tends to keep blood in it. However, despite these inhibitory
influences, there is some regurgitation of blood into the veins.
VENTRICULAR SYSTOLE
At the start of ventricular systole, the AV valves close. Ventricular muscle
initially shortens relatively little, but intraventricular pressure rises
sharply as the myocardium presses on the blood in the ventricle. This period of
isovolumetric (isovolumic, isometric) ventricular contraction lasts
about 0.05 s, until the pressures in the left and right ventricles exceed the
pressures in the aorta (80 mm Hg; 10.6 kPa) and pulmonary artery (10 mm Hg) and
the aortic and pulmonary valves open. During isovolumetric contraction, the AV
valves bulge into the atria, causing a small but sharp rise in atrial pressure.
When the aortic and pulmonary valves open, the phase of ventricular
ejection begins. Ejection is rapid at first, slowing down as systole
progresses. The intraventricular pressure rises to a maximum and then declines
somewhat before ventricular systole ends. Peak pressures in the left and right
ventricles are about 120 and 25 mm Hg, respectively. Late in systole, pressure
in the aorta actually exceeds that in the left ventricle, but for a short period
momentum keeps the blood moving forward. The AV valves are pulled down by the
contractions of the ventricular muscle, and atrial pressure drops. The amount of
blood ejected by each ventricle per stroke at rest is 70–90 mL. The
end-diastolic ventricular volume is about 130 mL. Thus, about 50 mL of blood
remains in each ventricle at the end of systole (end-systolic ventricular
volume), and the ejection fraction, the percentage of the
end-diastolic ventricular volume that is ejected with each stroke, is about
65%. The ejection fraction is a valuable index of ventricular function. It can
be measured by injecting radionuclide-labeled red blood cells and imaging the
cardiac blood pool at the end of diastole and the end of systole (equilibrium
radionuclide angiocardiography), or by computed tomography.
EARLY DIASTOLE
Once the ventricular muscle is fully contracted, the already falling ventricular
pressures drop more rapidly. This is the period of protodiastole, which
lasts about 0.04 s. It ends when the momentum of the ejected blood is overcome
and the aortic and pulmonary valves close, setting up transient vibrations in
the blood and blood vessel walls. After the valves are closed, pressure
continues to drop rapidly during the period of isovolumetric ventricular
relaxation. Isovolumetric relaxation ends when the ventricular pressure
falls below the atrial pressure and the AV valves open, permitting the
ventricles to fill. Filling is rapid at first, then slows as the next cardiac
contraction approaches. Atrial pressure continues to rise after the end of
ventricular systole until the AV valves open, then drops and slowly rises again
until the next atrial systole.
Events of the cardiac cycle at a heart rate of 75 beats/min.
The phases of the cardiac cycle identified by the numbers at the bottom are as
follows: 1, atrial systole; 2, isovolumetric ventricular contraction; 3,
ventricular ejection; 4, isovolumetric ventricular relaxation; 5, ventricular
filling. Note that late in systole, aortic pressure actually exceeds left
ventricular pressure. However, the momentum of the blood keeps it flowing out of
the ventricle for a short period. The pressure relationships in the right
ventricle and pulmonary artery are similar. Atr. syst., atrial systole; ventric.
syst., ventricular systole.
Cardiac cycle is the sequence of electrical and mechanical events that repeats
with every heartbeat. There are two
phases namely diastolic phase and systolic phase.
TIMING
Although events on the two sides of the heart are similar, they are somewhat
asynchronous. Right atrial systole precedes left atrial systole, and contraction
of the right ventricle starts after that of the left. However, since pulmonary
arterial pressure is lower than aortic pressure, right ventricular ejection
begins before that of the left. During expiration, the pulmonary and aortic
valves close at the same time; but during inspiration, the aortic valve closes
slightly before the pulmonary. The slower closure of the pulmonary valve is due
to lower impedance of the pulmonary vascular tree. When measured over a period
of minutes, the outputs of the two ventricles are, of course, equal, but
transient differences in output during the respiratory cycle occur in normal
individuals.
LENGTH OF SYSTOLE AND DIASTOLE
Cardiac muscle has the unique property of contracting and repolarizing faster
when the heart rate is high (see Chapter 5), and the duration of systole
decreases from 0.27 s at a heart rate of 65 beats/min to 0.16 s at a rate of 200
beats/min. The reduced time interval is mainly due to a
decrease in the duration of systolic ejection. However, the duration of systole
is much more fixed than that of diastole, and when the heart rate is increased,
diastole is shortened to a much greater degree. For example, at a heart rate of
65 beats/min, the duration of diastole is 0.62 s, whereas at a heart rate of 200
beats/min, it is only 0.14 s. This fact has important physiologic and clinical
implications. It is during diastole that the heart muscle rests, and coronary
blood flow to the subendocardial portions of the left ventricle occurs only
during diastole. Furthermore, most of the ventricular filling occurs in
diastole. At heart rates up to about 180 beats/min, filling is adequate as long
as there is ample venous return, and cardiac output per minute is increased by
an increase in rate. However, at very high heart rates, filling may be
compromised to such a degree that cardiac output per minute falls.
Because it has a prolonged action potential, cardiac muscle cannot contract in
response to a second stimulus until near the end of the initial contraction.
Therefore, cardiac muscle cannot be tetanized like skeletal muscle. The highest
rate at which the ventricles can contract is theoretically about 400/min, but
in adults the AV node will not conduct more than about 230 impulses/min because
of its long refractory period. A ventricular contraction rate of more than
230/min is seen only in paroxysmal ventricular tachycardia.
Exact measurement of the duration of isovolumetric ventricular contraction is
difficult in clinical situations, but it is relatively easy to measure the
duration of total electromechanical systole (QS2), the
preejection period (PEP), and the left ventricular ejection time (LVET)
by recording the ECG, phonocardiogram, and carotid pulse simultaneously. QS2
is the period from the onset of the QRS complex to the closure of the aortic
valves, as determined by the onset of the second heart sound. LVET is the period
from the beginning of the carotid pressure rise to the dicrotic notch (see
below). PEP is the difference between QS2 and LVET and represents the time for
the electrical as well as the mechanical events that precede systolic ejection.
The ratio PEP/LVET is normally about 0.35, and it increases without a change in
QS2 when left ventricular performance is compromised in a variety of cardiac
diseases.
ARTERIAL PULSE
The blood forced into the aorta during systole not only moves the blood in the
vessels forward but also sets up a pressure wave that travels along the
arteries. The pressure wave expands the arterial walls as it travels, and the
expansion is palpable as the pulse. The rate at which the wave travels,
which is independent of and much higher than the velocity of blood flow, is
about 4 m/s in the aorta, 8 m/s in the large arteries, and 16 m/s in the small
arteries of young adults. Consequently, the pulse is felt in the radial artery
at the wrist about 0.1 s after the peak of systolic ejection into the aorta.
With advancing age, the arteries become more rigid, and the pulse wave moves
faster.
The strength of the pulse is determined by the pulse pressure and bears little
relation to the mean pressure. The pulse is weak (“thready”) in shock. It is
strong when stroke volume is large; for example, during exercise or after the
administration of histamine. When the pulse pressure is high, the pulse waves
may be large enough to be felt or even heard by the individual (palpitation,
“pounding heart”). When the aortic valve is incompetent (aortic regurgitation),
the pulse is particularly strong, and the force of systolic ejection may be
sufficient to make the head nod with each heartbeat. The pulse in aortic
regurgitation is called a Corrigan or water-hammer pulse.
The dicrotic notch, a small oscillation on the falling phase
of the pulse wave caused by vibrations set up when the aortic valve snaps shut,
is visible if the pressure wave is recorded but is not palpable at the wrist.
The pulmonary artery pressure curve also has a dicrotic notch produced by the
closure of the pulmonary valves.
ATRIAL PRESSURE CHANGES AND THE JUGULAR PULSE
Atrial pressure rises during atrial systole and continues to rise during
isovolumetric ventricular contraction when the AV valves bulge into the atria.
When the AV valves are pulled down by the contracting ventricular muscle,
pressure falls rapidly and then rises as blood flows into the atria until the
AV valves open early in diastole. The return of the AV valves to their relaxed
position also contributes to this pressure rise by reducing atrial capacity. The
atrial pressure changes are transmitted to the great veins, producing three
characteristic waves in the record of jugular pressure. The a wave is due
to atrial systole. As noted above, some blood regurgitates into the great veins
when the atria contract. In addition, venous inflow stops, and the resultant
rise in venous pressure contributes to the a
wave. The c wave is the transmitted manifestation of the rise in atrial
pressure produced by the bulging of the tricuspid valve into the atria during
isovolumetric ventricular contraction. The v wave mirrors the rise in
atrial pressure before the tricuspid valve opens during diastole. The jugular
pulse waves are superimposed on the respiratory fluctuations in venous pressure.
Venous pressure falls during inspiration as a result of the increased negative
intrathoracic pressure and rises again during expiration.
HEART SOUNDS
Two sounds are normally heard through a stethoscope during each cardiac cycle.
The first is a low, slightly prolonged “lub” (first sound), caused by
vibrations set up by the sudden closure of the AV valves at the start of
ventricular systole. The second is a shorter, high-pitched “dup” (second
sound), caused by vibrations associated with closure of the aortic and
pulmonary valves just after the end of ventricular systole. A soft, low-pitched
third sound is heard about one-third of the way through diastole in many
normal young individuals. It coincides with the period of rapid ventricular
filling and is probably due to vibrations set up by the inrush of blood. A
fourth sound can sometimes be heard immediately before the first sound when
atrial pressure is high or the ventricle is stiff in conditions such as
ventricular hypertrophy. It is due to ventricular filling and is rarely heard in
normal adults. The first sound has a duration of about 0.15 s and a frequency of
25–45 Hz. It is soft when the heart rate is low, because the ventricles are well
filled with blood and the leaflets of the AV valves float together before
systole. The second sound lasts about 0.12 s, with a frequency of 50 Hz. It is
loud and sharp when the diastolic pressure in the aorta or pulmonary artery is
elevated, causing the respective valves to shut briskly at the end of systole.
The interval between aortic and pulmonary valve closure during inspiration is
frequently long enough for the second sound to be reduplicated (physiologic
splitting of the second sound). Splitting also occurs in various diseases. The
third sound, when present, has a duration of 0.1 s.
MURMURS
Murmurs,
or bruits, are abnormal sounds heard in various parts of the vascular
system. The two terms are used interchangeably, though “murmur” is more
commonly used to denote noise heard over the heart than over blood vessels.
Blood flow is laminar, nonturbulent, and silent up to a critical velocity;
above this velocity (such as beyond an obstruction), blood flow is turbulent
and creates sounds. Blood flow speeds up when an artery or a heart valve is
narrowed. Examples of vascular
sounds outside the heart are the bruit heard over a large, highly vascular
goiter, the bruit heard over a carotid artery when its lumen is narrowed and
distorted by atherosclerosis, and the murmurs heard over an aneurysmal dilation
of one of the large arteries, an arteriovenous (A-V) fistula, or a patent ductus
arteriosus. The major—but certainly not the only—cause of cardiac murmurs is
disease of the heart valves. When the orifice of a valve is narrowed
(stenosis), blood flow through it is accelerated and turbulent. When a
valve is incompetent, blood flows through it backward (regurgitation),
again through a narrow orifice that accelerates flow. The timing (systolic or
diastolic) of a murmur due to any particular valve
can be predicted from a knowledge of the mechanical events of the cardiac cycle.
Murmurs due to disease of a particular valve can generally be heard best when
the stethoscope is directly over the valve. There are also other aspects of the
duration, character, accentuation, and transmission of the sound that help
locate its origin in one valve or another. One of the loudest murmurs is that
produced when blood flows backward in diastole through a hole in a cusp of the
aortic valve. Most murmurs can be heard only with the aid of the stethoscope,
but this high-pitched musical diastolic murmur is sometimes audible to the
unaided ear several feet from the patient. In patients with congenital
interventricular septal defects, flow from the left to the right ventricle
causes a systolic murmur. Soft murmurs may also be heard in patients with
interatrial septal defects, although they are not a constant finding. Soft
systolic murmurs are also common in individuals, especially children, who have
no cardiac disease. Systolic murmurs are also heard in anemic patients as a
result of the low viscosity of the blood and associated rapid flow.
Heartbeat Coordination
The heart is a dual pump in that the left and right sides of the heart pump
blood separately—but simultaneously—into the systemic and pulmonary vessels.
Efficient pumping of blood requires that the atria contract first, followed
almost immediately by the ventricles. Contraction of cardiac muscle, like that
of skeletal muscle and many smooth muscles, is triggered by depolarization of
the plasma membrane. Gap junctions interconnect myocardial cells and allow
action potentials to spread from one cell to another. The initial excitation of
one cardiac cell therefore eventually results in the excitation of all cardiac
cells. This initial depolarization normally arises in a small group of
conducting-system cells called the sinoatrial (SA) node,
located in the right atrium near the entrance of the superior vena cava. The
action potential then spreads from the SA node throughout the atria and then
into and throughout the ventricles.
Sequence of Excitation
The SA node is normally the pacemaker for the entire heart. Its depolarization
generates the action potential that leads to depolarization of all other cardiac
muscle cells. As we will see later, electrical excitation of the heart is
coupled with contraction of cardiac muscle. Therefore, the discharge rate of the
SA node determines the heart rate, the number of times the heart
contracts per minute. The action
potential initiated in the SA node spreads throughout the myocardium, passing
from cell to cell by way of gap junctions. Depolarization first spreads through
the muscle cells of the atria, with conduction rapid enough that the right and
left atria contract at essentially the same time.
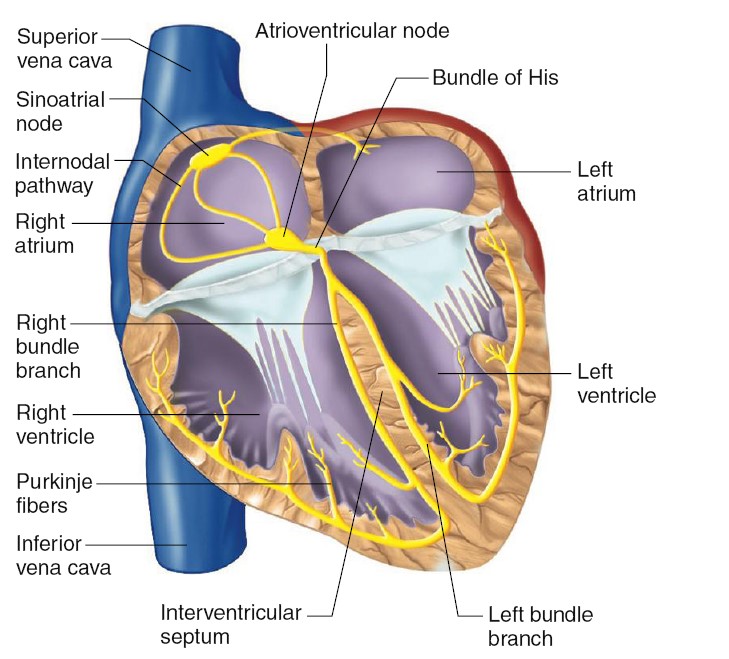
The spread of the action potential to the ventricles involves a more complicated
conducting system,
which consists of modified cardiac cells that have lost contractile capability
but that conduct action potentials with low resistance. The link between atrial
depolarization and ventricular depolarization is a portion of the conducting
system called the atrioventricular (AV) node, located at
the base of the right atrium. The action potential is conducted relatively
rapidly from the SA node to the AV node through internodal pathways. The
AV node is an elongated structure with a particularly important characteristic:
The propagation of action potentials through the AV node is relatively slow
(requiring approximately 0.1 sec). This delay allows atrial contraction to
be completed before ventricular excitation occurs.
After the AV node has become excited, the action potential propagates down the
interventricular septum. This pathway has conducting-system fibers called the
bundle of His (pronounced “hiss”), or atrioventricular bundle. The fibers
were identified by Wilhelm His in 1893. The AV node and the bundle of His
constitute the only electrical connection between the atria and the ventricles.
Except for this pathway, the atria are separated from the ventricles by a layer
of nonconducting connective tissue. Within the interventricular septum, the
bundle of His divides into right and left bundle branches, which separate
at the bottom (apex) of the heart and enter the walls of both ventricles. These
pathways are composed of Purkinje fibers, which are largediameter,
rapidly conducting cells connected by low-resistance gap junctions. The
branching network of Purkinje fibers conducts the action potential rapidly to
myocytes throughout the ventricles. The rapid conduction along the Purkinje
fibers and the diffuse distribution of these fibers cause depolarization of all
right and left ventricular cells to occur nearly simultaneously and ensure a
single coordinated contraction. Actually, though, depolarization and contraction
do begin slightly earlier in the apex of the ventricles and then spread upward.
The result is an efficient contraction that moves blood toward the exit valves,
like squeezing a tube of toothpaste from the bottom up.
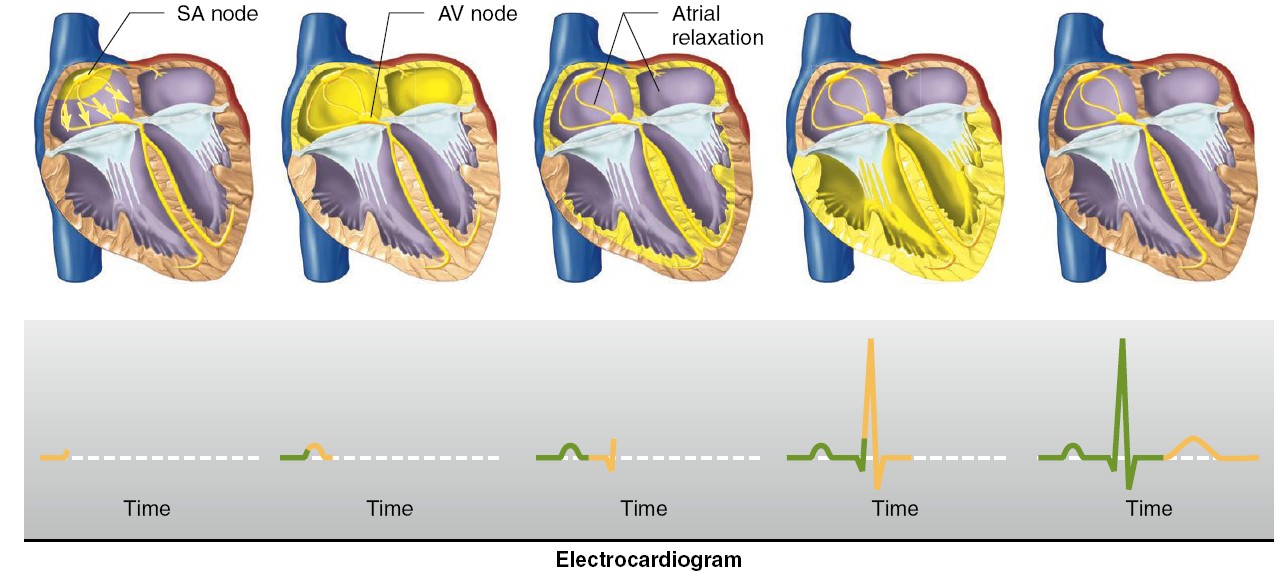
THE SINUS NODE IS THE NORMAL PACEMAKER OF THE HEART
In the discussion thus far of the genesis and transmission of the cardiac
impulse through the heart, we have noted that the impulse normally arises in the
sinus node. In some abnormal conditions, this is not the case. Other parts of
the heart can also exhibit intrinsic rhythmical excitation in the same way that
the sinus nodal fibers do; this capability is particularly true of the A-V nodal
and Purkinje fibers. The A-V nodal fibers, when not stimulated from some outside
source, discharge at an intrinsic rhythmical rate of 40 to 60 times per minute,
and the Purkinje fibers discharge at a rate somewhere between 15 and 40 times
per minute. These rates are in contrast to the normal rate of the sinus node of
70 to 80 times per minute.
The discharge rate of the sinus node is considerably faster than the natural
self-excitatory discharge rate of either the A-V node or the Purkinje fibers.
Purkinje J.E., modified muscle fiber in subendothelial region in 1839. Each time
the sinus node discharges, its impulse is conducted into both the A-V node and
the Purkinje fibers, also discharging their excitable membranes. However, the
sinus node discharges again before either the A-V node or the Purkinje fibers
can reach their own thresholds for self-excitation. Therefore, the new impulse
from the sinus node discharges both the A-V node and the Purkinje fibers before
self-excitation can occur in either of these sites. Thus, the sinus node
controls the beat of the heart because its rate of rhythmical discharge is
faster than that of any other part of the heart. Therefore, the sinus node is
almost always the pacemaker of the normal heart.
Abnormal Pacemakers -“Ectopic” Pacemaker.
Occasionally some other part of the heart develops a rhythmical discharge rate
that is more rapid than that of the sinus node. For instance, this development
sometimes occurs in the A-V node or in the Purkinje fibers when one of these
becomes abnormal. In either case, the pacemaker of the heart shifts from the
sinus node to the A-V node or to the excited Purkinje fibers. Under rarer
conditions, a place in the atrial or ventricular muscle develops excessive
excitability and becomes the pacemaker.
A pacemaker elsewhere than the sinus node is called an “ectopic” pacemaker.
An ectopic pacemaker causes an abnormal sequence of contraction of the different
parts of the heart and can cause significant debility of heart pumping. Another
cause of shift of the pacemaker is blockage of transmission of the cardiac
impulse from the sinus node to the other parts of the heart. The new pacemaker
then occurs most frequently at the A-V node or in the penetrating portion of the
A-V bundle on the way to the ventricles. When A-V block occurs—that is, when the
cardiac impulse fails to pass from the atria into the ventricles through the A-V
nodal and bundle system—the atria continue to beat at the normal rate of rhythm
of the sinus node, while a new pacemaker usually develops in the Purkinje system
of the ventricles and drives the ventricular muscle at a new rate somewhere
between 15 and 40 beats per minute. After sudden A-V bundle block, the Purkinje
system does not begin to emit its intrinsic rhythmical impulses until 5 to 20
seconds later because, before the blockage, the Purkinje fibers had been
“overdriven” by the rapid sinus impulses and, consequently, are in a suppressed
state. During these 5 to 20 seconds, the ventricles fail to pump blood, and the
person faints after the first 4 to 5 seconds because of lack of blood flow to
the brain. This delayed pickup of the heartbeat is called Stokes-Adams
syndrome. If the delay period is too long, it can lead to death Robert Adam
and William Stokes identified the syndrome.
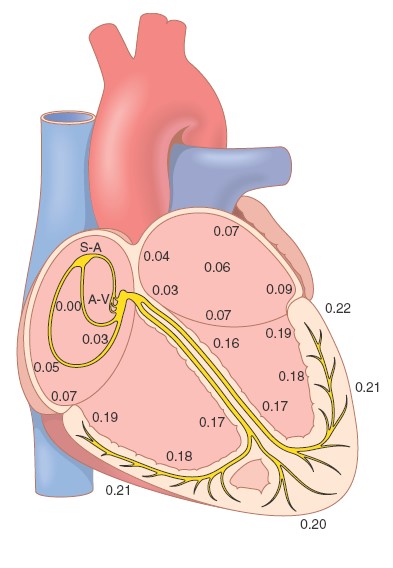
Transmission of the cardiac impulse through the heart, showing the time of
appearance (in fractions of a second after initial appearance at the sinoatrial
node) in different parts of the heart. A-V, atrioventricular; S-A, sinoatrial.
The Electrocardiogram
The body is a good conductor of electricity because tissue fluids have a high
concentration of ions that move (creating a current) in response to potential
differences. Potential differences generated by the heart are conducted to the
body surface, where they can be recorded by surface electrodes placed on the
skin. The recording thus obtained is called an electrocardiogram ( ECG
or EKG ); the recording device is called an electrocardiograph.
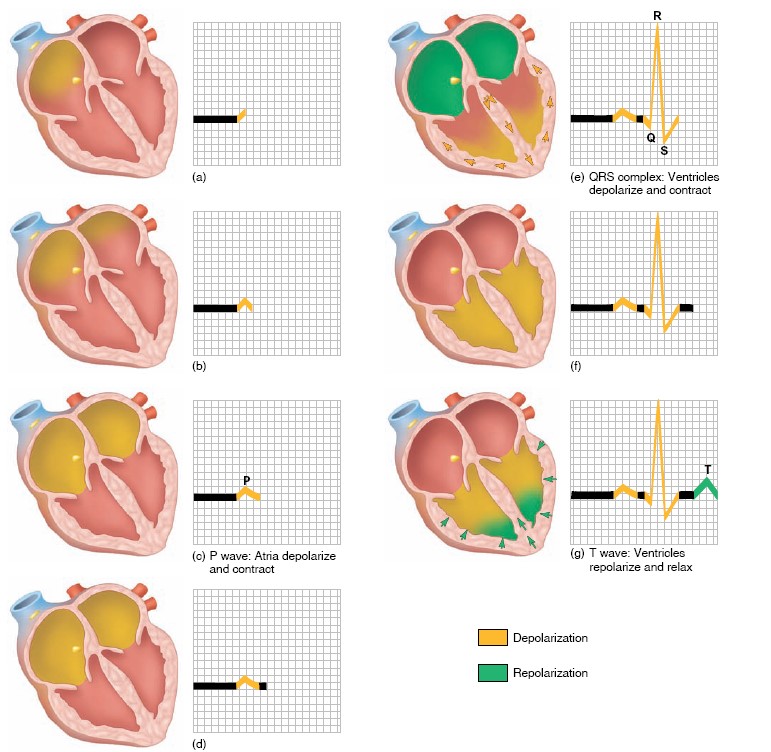
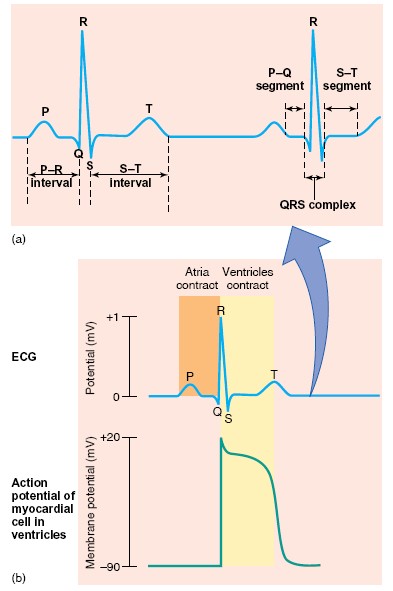
Each cardiac cycle produces three distinct ECG waves, designated P, QRS,
and T. Note that the ECG is not a recording of action potentials, but
it does result from the production and conduction of action potentials in the
heart. The correlation of an action potential produced in the ventricles to the
waves of the ECG. This figure shows that the spread of depolarization through
the ventricles (indicated by the QRS, described shortly) corresponds to the
action potential, and thus to contraction of the ventricles. The spread of
depolarization through the atria causes a potential difference that is indicated
by an upward deflection of the ECG line. When about half the mass of the atria
is depolarized, this upward deflection reaches a maximum value because the
potential difference between the depolarized and unstimulated portions of the
atria is at a maximum. When the entire mass of the atria is depolarized, the ECG
returns to baseline because all regions of the atria have the same polarity. The
spread of atrial depolarization thereby creates the P wave. Conduction of
the impulse into the ventricles similarly creates a potential difference that
results in a sharp upward deflection of the ECG line, which then returns to the
baseline as the entire mass of the ventricles becomes depolarized. The spread of
the depolarization into the ventricles is thereby represented by the QRS
wave. The plateau phase of the cardiac action potential is related to the
S-T segment of the ECG. Finally, repolarization of the ventricles produces
the T wave. You might be surprised that ventricular depolarization (the
QRS wave) and repolarization (the T wave) point in the same direction, although
they are produced by opposite potential changes. This is because depolarization
of the ventricles occurs from endocardium to epicardium, whereas repolarization
spreads in the opposite direction, from epicardium to endocardium. There are two
types of ECG recording electrodes, or “leads.” The bipolar limb leads
record the voltage between electrodes placed on the wrists and legs. These
bipolar leads include lead I (right arm to left arm), lead II (right arm to left
leg), and lead III (left arm to left leg). The right leg is used as a ground
lead. In the unipolar leads, voltage is recorded between a single
“exploratory electrode” placed on the body and an electrode that is built into
the electrocardiograph and maintained at zero potential (ground).
The unipolar limb leads are placed on the right arm, left arm, and left leg, and
are abbreviated AVR, AVL, and AVF, respectively.
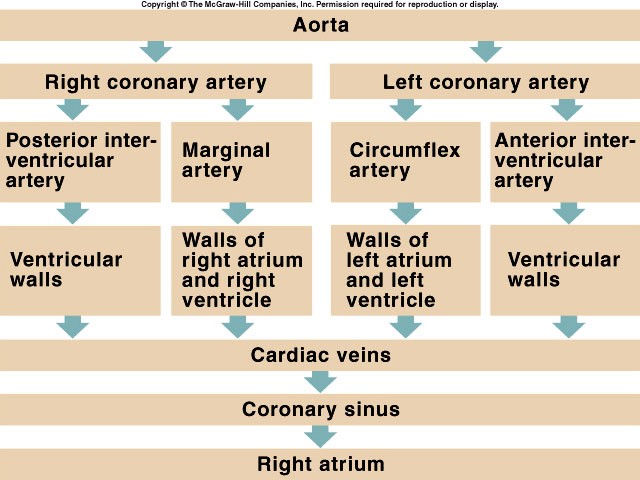
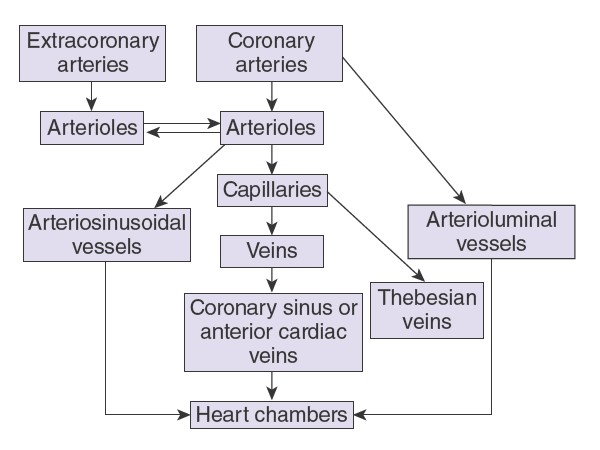
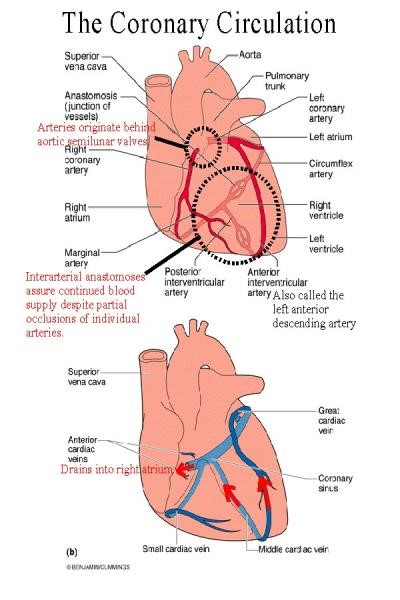
CORONARY CIRCULATION
The two coronary arteries that supply the myocardium arise from the sinuses
behind two of the cusps of the aortic valve at the root of the aorta. Eddy
currents keep the valves away from the orifices of the arteries, and they are
patent throughout the cardiac cycle. Most of the venous blood returns to the
heart through the coronary sinus and anterior cardiac veins, which drain into
the right atrium. In addition, there are other vessels that empty directly into
the heart chambers. These include arteriosinusoidal vessels, sinusoidal
capillary-like vessels that connect arterioles to the chambers; thebesian
veins that connect capillaries to the chambers; and a few arterioluminal
vessels that are small arteries draining directly into the chambers. A few
anastomoses occur between the coronary arterioles and extracardiac arterioles,
especially around the mouths of the great veins. Anastomoses between coronary
arterioles in humans only pass particles less than 40 μm in diameter, but
evidence indicates that these channels enlarge and increase in number in
patients with coronary artery disease.