CANCER
Cancer is a particularly appropriate topic for the concluding chapter of this book because it results from a breakdown of the regulatory mechanisms that govern normal cell behavior. As discussed in preceding chapters, the proliferation, differentiation, and survival of individual cells in multicellular organisms are carefully regulated to meet the needs of the organism as a whole. This regulation is lost in cancer cells, which grow and divide in an uncontrolled manner, ultimately spreading throughout the body and interfering with the function of normal tissues and organs.
Because cancer results from defects in fundamental cell regulatory mechanisms, it is a disease that ultimately has to be understood at the molecular and cellular levels. Indeed, understanding cancer has been an objective of molecular and cellular biologists for many years. Importantly, studies of cancer cells have also illuminated the mechanisms that regulate normal cell behavior. In fact, many of the proteins that play key roles in cell signaling, regulation of the cell cycle, and control of programmed cell death were first identified because abnormalities in their activities led to the uncontrolled proliferation of cancer cells. The study of cancer has thus contributed significantly to our understanding of normal cell regulation, as well as vice versa.
The Development and Causes of Cancer
The fundamental abnormality resulting in the development of cancer is the continual unregulated proliferation of cancer cells. Rather than responding appropriately to the signals that control normal cell behavior, cancer cells grow and divide in an uncontrolled manner, invading normal tissues and organs and eventually spreading throughout the body. The generalized loss of growth control exhibited by cancer cells is the net result of accumulated abnormalities in multiple cell regulatory systems and is reflected in several aspects of cell behavior that distinguish cancer cells from their normal counterparts.
Types of Cancer
Cancer can result from abnormal proliferation of any of the different kinds of cells in the body, so there are more than a hundred distinct types of cancer, which can vary substantially in their behavior and response to treatment. The most important issue in cancer pathology is the distinction between benign and malignant tumors. A tumor is any abnormal proliferation of cells, which may be either benign or malignant. A benign tumor, such as a common skin wart, remains confined to its original location, neither invading neither surrounding normal tissue nor spreading to distant body sites. A malignant tumor, however, is capable of both invading surrounding normal tissue and spreading throughout the body via the circulatory or lymphatic systems (metastasis). Only malignant tumors are properly referred to as cancers, and it is their ability to invade and metastasize that makes cancer so dangerous. Whereas benign tumors can usually be removed surgically, the spread of malignant tumors to distant body sites frequently makes them resistant to such localized treatment.
Both benign and malignant tumors are classified according to the type of cell from which they arise. Most cancers fall into one of three main groups: carcinomas, sarcomas, and leukemias or lymphomas. Carcinomas, which include approximately 90% of human cancers, are malignancies of epithelial cells. Sarcomas, which are rare in humans, are solid tumors of connective tissues, such as muscle, bone, cartilage, and fibrous tissue. Leukemias and lymphomas, which account for approximately 8% of human malignancies, arise from the blood-forming cells and from cells of the immune system, respectively. Tumors are further classified according to tissue of origin (e.g., lung or breast carcinomas) and the type of cell involved. For example, fibrosarcomas arise from fibroblasts, and erythroid leukemias from precursors of erythrocytes (red blood cells).
Although there are many kinds of cancer, only a few occur frequently. More than a million cases of cancer are diagnosed annually in the United States, and more than 500,000 Americans die of cancer each year. Cancers of 10 different body sites account for more than 75% of this total cancer incidence. The four most common cancers, accounting for more than half of all cancer cases, are those of the breast, prostate, lung, and colon/rectum. Lung cancer, by far the most lethal, is responsible for nearly 30% of all cancer deaths.
The Development of Cancer
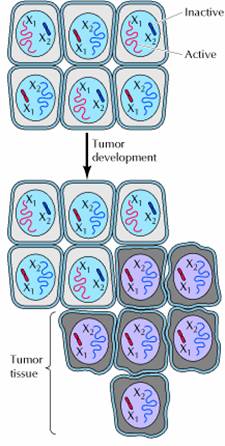
One of the fundamental features of cancer is tumor clonality, the development of tumors from single cells that begin to proliferate abnormally. The single-cell origin of many tumors has been demonstrated by analysis of X chromosome inactivation. One member of the X chromosome pair is inactivated by being converted to heterochromatin in female cells. X inactivation occurs randomly during embryonic development, so one X chromosome is inactivated in some cells, while the other X chromosome is inactivated in other cells. Thus, if a female is heterozygous for an X chromosome gene, different alleles will be expressed in different cells. Normal tissues are composed of mixtures of cells with different inactive X chromosomes, so expression of both alleles is detected in normal tissues of heterozygous females. In contrast, tumor tissues generally express only one allele of a heterozygous X chromosome gene. The implication is that all of the cells constituting such a tumor were derived from a single cell of origin, in which the pattern of X inactivation was fixed before the tumor began to develop.
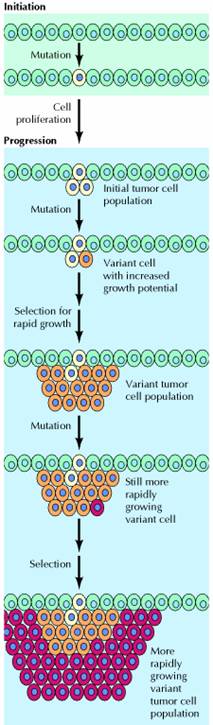
The clonal origin of tumors does not, however, imply that the original progenitor cell that gives rise to a tumor has initially acquired all of the characteristics of a cancer cell. On the contrary, the development of cancer is a multistep process in which cells gradually become malignant through a progressive series of alterations. One indication of the multistep development of cancer is that most cancers develop late in life. The incidence of colon cancer, for example, increases more than tenfold between the ages of 30 and 50, and another tenfold between 50 and 70. Such a dramatic increase of cancer incidence with age suggests that most cancers develop as a consequence of multiple abnormalities, which accumulate over periods of many years.
At the cellular level, the development of cancer is viewed as a multistep process involving mutation and selection for cells with progressively increasing capacity for proliferation, survival, invasion, and metastasis. The first step in the process, tumor initiation, is thought to be the result of a genetic alteration leading to abnormal proliferation of a single cell. Cell proliferation then leads to the outgrowth of a population of clonally derived tumor cells. Tumor progression continues as additional mutations occur within cells of the tumor population. Some of these mutations confer a selective advantage to the cell, such as more rapid growth, and the descendants of a cell bearing such a mutation will consequently become dominant within the tumor population. The process is called clonal selection, since a new clone of tumor cells has evolved on the basis of its increased growth rate or other properties (such as survival, invasion, or metastasis) that confer a selective advantage. Clonal selection continues throughout tumor development, so tumors continuously become more rapid-growing and increasingly malignant.
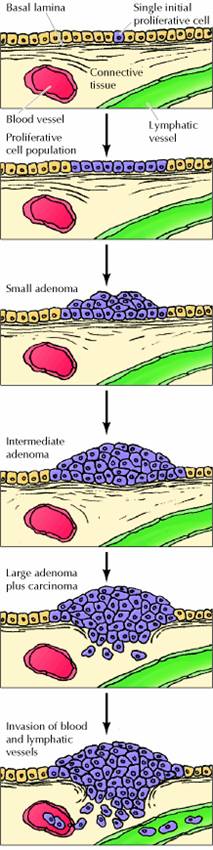
Studies of colon carcinomas have provided a clear example of tumor progression during the development of a common human malignancy. The earliest stage in tumor development is increased proliferation of colon epithelial cells. One of the cells within this proliferative cell population is then thought to give rise to a small benign neoplasm (an adenoma or polyp). Further rounds of clonal selection lead to the growth of adenomas of increasing size and proliferative potential.
Malignant carcinomas then arise from the benign adenomas, indicated by invasion of the tumor cells through the basal lamina into underlying connective tissue. The cancer cells then continue to proliferate and spread through the connective tissues of the colon wall. Eventually the cancer cells penetrate the wall of the colon and invade other abdominal organs, such as the bladder or small intestine. In addition, the cancer cells invade blood and lymphatic vessels, allowing them to metastasize throughout the body.
Causes of Cancer
Substances that cause cancer, called carcinogens, have been identified both by studies in experimental animals and by epidemiological analysis of cancer frequencies in human populations (e.g., the high incidence of lung cancer among cigarette smokers). Since the development of malignancy is a complex multistep process, many factors may affect the likelihood that cancer will develop, and it is overly simplistic to speak of single causes of most cancers. Nonetheless, many agents, including radiation, chemicals, and viruses, have been found to induce cancer in both experimental animals and humans.
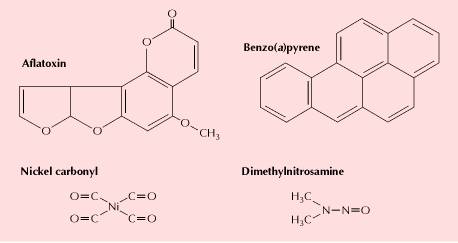
Radiation and many chemical carcinogens act by damaging DNA and inducing mutations. These carcinogens are generally referred to as initiating agents, since the induction of mutations in key target genes is thought to be the initial event leading to cancer development. Some of the initiating agents that contribute to human cancers include solar ultraviolet radiation (the major cause of skin cancer), carcinogenic chemicals in tobacco smoke, and aflatoxin (a potent liver carcinogen produced by some molds that contaminate improperly stored supplies of peanuts and other grains). The carcinogens in tobacco smoke (including benzo(a)pyrene, dimethylnitrosamine, and nickel compounds) are the major identified causes of human cancer. Smoking is the undisputed cause of 80 to 90% of lung cancers, as well as being implicated in cancers of the oral cavity, pharynx, larynx, esophagus, and other sites. In total, it is estimated that smoking is responsible for nearly one-third of all cancer deaths—an impressive toll for a single carcinogenic agent.
Other carcinogens contribute to cancer development by stimulating cell proliferation, rather than by inducing mutations. Such compounds are referred to as tumor promoters, since the increased cell division they induce is required for the outgrowth of a proliferative cell population during early stages of tumor development. The phorbol esters that stimulate cell proliferation by activating protein kinase C are classic examples. Their activity was defined by studies of chemical induction of skin tumors in mice. Tumorigenesis in this system can be initiated by a single treatment with a mutagenic carcinogen. Tumors do not develop, however, unless the mice are subsequently treated with a tumor promoter (usually a phorbol ester) to stimulate proliferation of the mutated cells.
Hormones, particularly estrogens, are important as tumor promoters in the development of some human cancers. The proliferation of cells of the uterine endometrium, for example, is stimulated by estrogen, and exposure to excess estrogen significantly increases the likelihood that a woman will develop endometrial cancer. The risk of endometrial cancer is therefore substantially increased by long-term postmenopausal estrogen replacement therapy with high doses of estrogen alone. Fortunately, this risk is minimized by administration of progesterone to counteract the stimulatory effect of estrogen on endometrial cell proliferation. However, long-term therapy with combinations of estrogen and progesterone may lead to an increased risk of breast cancer.
In addition to chemicals and radiation, some viruses induce cancer both in experimental animals and in humans. The common human cancers caused by viruses include liver cancer and cervical carcinoma, which together account for 10 to 20% of worldwide cancer incidence. These viruses are important not only as causes of human cancer; as discussed later in this chapter, studies of tumor viruses have played a key role in elucidating the molecular events responsible for the development of cancers induced by both viral and nonviral carcinogens.
Properties of Cancer Cells
The uncontrolled growth of cancer cells results from accumulated abnormalities affecting many of the cell regulatory mechanisms that have been discussed in preceding chapters. This relationship is reflected in several aspects of cell behavior that distinguish cancer cells from their normal counterparts. Cancer cells typically display abnormalities in the mechanisms that regulate normal cell proliferation, differentiation, and survival. Taken together, these characteristic properties of cancer cells provide a description of malignancy at the cellular level.
The uncontrolled proliferation of cancer cells in vivo is mimicked by their behavior in cell culture. A primary distinction between cancer cells and normal cells in culture is that normal cells display density-dependent inhibition of cell proliferation. Normal cells proliferate until they reach a finite cell density, which is determined in part by the availability of growth factors added to the culture medium (usually in the form of serum). They then cease proliferating and become quiescent, arrested in the G0 stage of the cell cycle. The proliferation of most cancer cells, however, is not sensitive to density-dependent inhibition. Rather than responding to the signals that cause normal cells to cease proliferation and enter G0, tumor cells generally continue growing to high cell densities in culture, mimicking their uncontrolled proliferation in vivo.
A related difference between normal cells and cancer cells is that many cancer cells have reduced requirements for extracellular growth factors. The proliferation of most cells is controlled, at least in part, by polypeptide growth factors. For some cell types, particularly fibroblasts, the availability of serum growth factors is the principal determinant of their proliferative capacity in culture. The growth factor requirements of these cells are closely related to the phenomenon of density-dependent inhibition, since the density at which normal fibroblasts become quiescent is proportional to the concentration of serum growth factors in the culture medium.
The growth factor requirements of many tumor cells are reduced compared to their normal counterparts, contributing to the unregulated proliferation of tumor cells both in vitro and in vivo. In some cases, cancer cells produce growth factors that stimulate their own proliferation. Such abnormal production of a growth factor by a responsive cell leads to continuous autostimulation of cell division (autocrine growth stimulation), and the cancer cells are therefore less dependent on growth factors from other, physiologically normal sources. In other cases, the reduced growth factor dependence of cancer cells results from abnormalities in intracellular signaling systems, such as unregulated activity of growth factor receptors or other proteins (e.g., Ras proteins or protein kinases) that act as elements of signal transduction pathways leading to cell proliferation.
Cancer cells are also less stringently regulated than normal cells by cell-cell and cell-matrix interactions. Most cancer cells are less adhesive than normal cells, often as a result of reduced expression of cell surface adhesion molecules. For example, loss of E-cadherin, the principal adhesion molecule of epithelial cells, is important in the development of carcinomas (epithelial cancers). As a result of reduced expression of cell adhesion molecules, cancer cells are comparatively unrestrained by interactions with other cells and tissue components, contributing to the ability of malignant cells to invade and metastasize. The reduced adhesiveness of cancer cells also results in morphological and cytoskeletal alterations: Many tumor cells are rounder than normal, in part because they are less firmly attached to either the extracellular matrix or neighboring cells.
A striking difference in the cell-cell interactions displayed by normal cells and those of cancer cells is illustrated by the phenomenon of contact inhibition. Normal fibroblasts migrate across the surface of a culture dish until they make contact with a neighboring cell. Further cell migration is then inhibited, and normal cells adhere to each other, forming an orderly array of cells on the culture dish surface. Tumor cells, in contrast, continue moving after contact with their neighbors, migrating over adjacent cells, and growing in disordered, multilayered patterns. Not only the movement but also the proliferation of many normal cells is inhibited by cell-cell contact, and cancer cells are characteristically insensitive to such contact inhibition of growth.
Two additional properties of cancer cells affect their interactions with other tissue components, thereby playing important roles in invasion and metastasis. First, malignant cells generally secrete proteases that digest extracellular matrix components, allowing the cancer cells to invade adjacent normal tissues. Secretion of collagenase, for example, appears to be an important determinant of the ability of carcinomas to digest and penetrate through basal laminae to invade underlying connective tissue. Second, cancer cells secrete growth factors that promote the formation of new blood vessels (angiogenesis). Angiogenesis is needed to support the growth of a tumor beyond the size of about a million cells, at which point new blood vessels are required to supply oxygen and nutrients to the proliferating tumor cells. Such blood vessels are formed in response to growth factors, secreted by the tumor cells that stimulate proliferation of endothelial cells in the walls of capillaries in surrounding tissue, resulting in the outgrowth of new capillaries into the tumor. The formation of such new blood vessels is important not only in supporting tumor growth, but also in metastasis. The actively growing new capillaries formed in response to angiogenic stimulation are easily penetrated by the tumor cells, providing a ready opportunity for cancer cells to enter the circulatory system and begin the metastatic process.
Another general characteristic of most cancer cells is that they fail to differentiate normally. Such defective differentiation is closely coupled to abnormal proliferation, since; most fully differentiated cells either cease cell division or divide only rarely. Rather than carrying out their normal differentiation program, cancer cells are usually blocked at an early stage of differentiation, consistent with their continued active proliferation.
The leukemias provide a particularly good example of the relationship between defective differentiation and malignancy. All of the different types of blood cells are derived from a common stem cell in the bone marrow. Descendants of these cells then become committed to specific differentiation pathways. Some cells, for example, differentiate to form erythrocytes whereas others differentiate to form lymphocytes, granulocytes, or macrophages. Cells of each of these types undergo several rounds of division as they differentiate, but once they become fully differentiated, cell division ceases. Leukemic cells, in contrast, fail to undergo terminal differentiation. Instead, they become arrested at early stages of maturation at which they retain their capacity for proliferation and continue to reproduce.
Programmed cell death, or apoptosis, is an integral part of the differentiation program of many cell types, including blood cells. Many cancer cells fail to undergo apoptosis, and therefore exhibit increased life spans compared to their normal counterparts. This failure of cancer cells to undergo programmed cell death contributes substantially to tumor development. For example, the survival of many normal cells is dependent on signals from growth factors or from the extracellular matrix that prevent apoptosis. In contrast, tumor cells are often able to survive in the absence of growth factors required by their normal counterparts. Such a failure of tumor cells to undergo apoptosis when deprived of normal environmental signals may be important not only in primary tumor development but also in the survival and growth of metastatic cells in abnormal tissue sites. Normal cells also undergo apoptosis following DNA damage, while many cancer cells fail to do so. In this case, the failure to undergo apoptosis contributes to the resistance of cancer cells to irradiation and many chemotherapeutic drugs, which act by damaging DNA. Abnormal cell survival, as well as cell proliferation, thus plays a major role in the unrelenting growth of cancer cells in an animal.
ONCOGENES
Since the early proposals of Boveri more than a century ago, much experimental evidence has confirmed that, at the molecular level, cancer is a result of lesions in the cellular deoxyribonucleic acid (DNA). First, it has been observed that a cancer cell transmits to its daughter cells the phenotypic features characterizing the cancerous state. Second, most of the recognized mutagenic compounds are also carcinogenic, having as a target cellular DNA. Finally, the karyotyping of several types of human tumors, particularly those belonging to the hematopoietic system, led to the identification of recurrent qualitative and numerical chromosomal aberrations, reflecting pathologic re-arrangements of the cellular genome. Taken together, these observations suggest that the molecular pathogenesis of human cancer is due to structural and/or functional alterations of specific genes whose normal function is to control cellular growth and differentiation or, in different terms, cell birth and cell death.
The identification and characterization of the genetic elements playing a role in the scenario of human cancer pathogenesis have been made possible by the development of DNA recombinant techniques during the last two decades. One milestone was the use of the DNA transfection technique that helped clarify the cellular origin of the “viral oncogenes.” The latter were previously characterized as the specific genetic elements capable of conferring the tumorigenic properties to the ribonucleic acid (RNA) tumor viruses also known as retroviruses. Furthermore, the transfection technique led to the identification of cellular transforming genes that do not have a viral counterpart. Besides the source of their original identification, viral or cellular genome, these transforming genetic elements have been designated as protooncogenes in their normal physiologic version and oncogenes when altered in cancer. A second relevant experimental approach has regarded the identification and characterization of clonal and recurrent cytogenetic abnormalities in cancer cells, especially those derived from the hematopoietic system. Several oncogenes have been thus defined by molecular cloning of the chromosomal breakpoints, including translocations and inversions. Additional oncogenes have been identified through the analysis of chromosomal regions anomalously stained (homogeneously staining regions), representing gene amplification. Finally, the detection of chromosome deletions has been instrumental in the process of identification and cloning of a second class of cancer-associated genes, the tumor suppressors. Contrary to the oncogenes that are activated by dominant mutations and whose activity is to promote cell growth, tumor suppressors act in the normal cell as negative controllers of cell growth and are inactive in tumor cells. In general, therefore, the mutations inactivating tumor suppressor genes are of the recessive type.
Recently, a third class of cancer-associated genes has been defined thanks to the analysis of tumors of a particular type; that is, tumors in which an inherited mutated predisposing gene plays a significant role. These tumors include cancers in patients suffering from hereditary nonpolyposis colorectal cancer syndromes.
The genes implicated in these tumors have been defined as mutator genes or genes involved in the DNA-mismatch repair process. Although not directly involved in the carcinogenesis process, these genes, when inactivated, expose the cells to a very high mutagenic load that eventually may involve the activation of oncogenes and the inactivation of tumor suppressors.
In this chapter, the methods by which oncogenes were discovered will be first described. The various functions of cellular protooncogenes will then be presented, and the genetic mechanisms of protooncogene activation will be summarized. Finally, the role of specific oncogenes in the initiation and progression of human tumors will be discussed.
Discovery and identification of oncogenes
The first oncogenes were discovered through the study of retroviruses, RNA tumor viruses whose genomes are reverse-transcribed into DNA in infected animal cells. During the course of infection, retroviral DNA is inserted into the chromosomes of host cells. The integrated retroviral DNA, called the provirus, replicates along with the cellular DNA of the host. Transcription of the DNA provirus leads to the production of viral progeny that bud through the host cell membrane to infect other cells. Two categories of retroviruses are classified by their time course of tumor formation in experimental animals. Acutely transforming retroviruses can rapidly cause tumors within days after injection. These retroviruses can also transform cell cultures to the neoplastic phenotype. Chronic or weakly oncogenic retroviruses can cause tissue-specific tumors in susceptible strains of experimental animals after a latency period of many months. Although weakly oncogenic retroviruses can replicate in vitro, these viruses do not transform cells in culture.
Retroviral oncogenes are altered versions of host cellular protooncogenes that have been incorporated into the retroviral genome by recombination with host DNA, a process known as retroviral transduction. This surprising discovery was made through study of the Rous sarcoma virus (RSV). RSV is an acutely transforming retrovirus first isolated from a chicken sarcoma over 80 years ago by Peyton Rous. Studies of RSV mutants in the early 1970s revealed that the transforming gene of RSV was not required for viral replication. Molecular hybridization studies then showed that the RSV transforming gene (designated v-src) was homologous to a host cellular gene (c-src) that was widely conserved in eukaryotic species. Studies of many other acutely transforming retroviruses from fowl, rodent, feline, and nonhuman primate species have led to the discovery of dozens of different retroviral oncogenes. In every case, these retroviral oncogenes are derived from normal cellular genes captured from the genome of the host. Viral oncogenes are responsible for the rapid tumor formation and efficient in vitro transformation activity characteristic of acutely transforming retroviruses.
In contrast to acutely transforming retroviruses, weakly oncogenic retroviruses do not carry viral oncogenes. These retroviruses, which include mouse mammary tumor virus (MMTV) and various animal leukemia viruses, induce tumors by a process called insertional mutagenesis. This process results from integration of the DNA provirus into the host genome in infected cells. In rare cells, the provirus inserts near a protooncogene. Expression of the protooncogene is then abnormally driven by the transcriptional regulatory elements contained within the long terminal repeats of the provirus. In these cases, proviral integration represents a mutagenic event that activates a protooncogene. Activation of the protooncogene then results in transformation of the cell, which can grow clonally into a tumor. The long latent period of tumor formation of weakly oncogenic retroviruses is therefore due to the rarity of the provirus insertional event that leads to tumor development from a single transformed cell. Insertional mutagenesis by weakly oncogenic retroviruses, first demonstrated in bursal lymphomas of chickens, frequently involves the same oncogenes (such as myc, myb, and erb B) that are carried by acutely transforming retroviruses. In many cases, however, insertional mutagenesis has been used as a tool to identify new oncogenes, including int-1, int-2, pim-1, and lck.
The demonstration of activated protooncogenes in human tumors was first shown by the DNA-mediated transformation technique. This technique, also called gene transfer or transfection assay, verifies the ability of donor DNA from a tumor to transform a recipient strain of rodent cells called NIH 3T3, an immortalized mouse cell line. This sensitive assay, which can detect the presence of single-copy oncogenes in a tumor sample, also enables the isolation of the transforming oncogene by molecular cloning techniques. After serial growth of the transformed NIH 3T3 cells, the human tumor oncogene can be cloned by its association with human repetitive DNA sequences. The first human oncogene isolated by the gene transfer technique was derived from a bladder carcinoma. Overall, approximately 20% of individual human tumors have been shown to induce transformation of NIH 3T3 cells in gene-transfer assays. The value of transfection assay was recently reinforced by the laboratory of Robert Weinberg, which showed that the ectopic expression of the telomerase catalytic subunit (hTERT), in combination with the simian virus 40 large T product and a mutated oncogenic H-ras protein, resulted in the direct tumorigenic conversion of normal human epithelial and fibroblast cells. Many of the oncogenes identified by gene-transfer studies are identical or closely related to those oncogenes transduced by retroviruses. Most prominent among these are members of the ras family that have been repeatedly isolated from various human tumors by gene transfer. A number of new oncogenes (such as neu, met, and trk) have also been identified by the gene-transfer technique. In many cases, however, oncogenes identified by gene transfer were shown to be activated by rearrangement during the experimental procedure and are not activated in the human tumors that served as the source of the donor DNA, as in the case of ret that was subsequently found genuinely rearranged and activated in papillary thyroid carcinomas.
Chromosomal translocations have served as guideposts for the discovery of many new oncogenes. Consistently recurring karyotypic abnormalities are found in many hematologic and solid tumors. These abnormalities include chromosomal rearrangements as well as the gain or loss of whole chromosomes or chromosome segments. The first consistent karyotypic abnormality identified in a human neoplasm was a characteristic small chromosome in the cells of patients with chronic myelogenous leukemia. Later identified as a derivative of chromosome 22, this abnormality was designated the Philadelphia chromosome, after its city of discovery. The application of chromosome banding techniques in the early 1970s enabled the precise cytogenetic characterization of many chromosomal translocations in human leukemia, lymphoma, and solid tumors. The subsequent development of molecular cloning techniques then enabled the identification of protooncogenes at or near chromosomal breakpoints in various neoplasms. Some of these protooncogenes, such as myc and abl, had been previously identified as retroviral oncogenes. In general, however, the cloning of chromosomal breakpoints has served as a rich source of discovery of new oncogenes involved in human cancer.
Oncogenes, protooncogenes, and their functions
Protooncogenes encode proteins that are involved in the control of cell growth. Alteration of the structure and/or expression of protooncogenes can activate them to become oncogenes capable of inducing in susceptible cells the neoplastic phenotype. Oncogenes can be classified into five groups based on the functional and biochemical properties of protein products of their normal counterparts (proto-oncogenes). These groups are (1) growth factors, (2) growth factor receptors, (3) signal transducers, (4) transcription factors, and (5) others, including programmed cell death regulators.
Growth Factors
Growth factors are secreted polypeptides that function as extracellular signals to stimulate the proliferation of target cells. Appropriate target cells must possess a specific receptor in order to respond to a specific type of growth factor. A well-characterized example is platelet-derived growth factor (PDGF), an approximately 30 kDa protein consisting of two polypeptide chains. PDGF is released from platelets during the process of blood coagulation. PDGF stimulates the proliferation of fibroblasts, a cell growth process that plays an important role in wound healing. Other well-characterized examples of growth factors include nerve growth factor, epidermal growth factor, and fibroblast growth factor.
The link between growth factors and retroviral oncogenes was revealed by study of the sis oncogene of simian sarcoma virus, a retrovirus first isolated from a monkey fibrosarcoma. Sequence analysis showed that sis encodes the beta chain of PDGF. This discovery established the principle that inappropriately expressed growth factors could function as oncogenes. Experiments demonstrated that the constitutive expression of the sis gene product (PDGF-β) was sufficient to cause neoplastic transformation of fibroblasts but not of cells that lacked the receptor for PDGF. Thus, transformation by sis requires interaction of the sis gene product with the PDGF receptor. The mechanism by which a growth factor affects the same cell that produces it is called autocrine stimulation. The constitutive expression of the sis gene product appears to cause neoplastic transformation by the mechanism of autocrine stimulation, resulting in self-sustained aberrant cell proliferation. This model, derived from experimental animal systems, has been recently demonstrated in a human tumor. Dermatofibrosarcoma protuberans (DP) is an infiltrative skin tumor that was demonstrated to present specific cytogenetic features: reciprocal translocation and supernumerary ring chromosomes, involving chromosomes 17 and 22. Molecular cloning of the breakpoints revealed a fusion between the collagen type Ia1 (COL1A1) gene and PDGF-β gene. The fusion gene resulted in a deletion of PDGF-β exon 1 and a constitutive release of this growth factor. Subsequent experiments of gene transfer of DPs genomic DNA into NIH 3T3 cells directly demonstrated the occurrence of an autocrine mechanism by the human rearranged PDGF-b gene involving the activation of the endogenous PDGF receptor. Another example of a growth factor that can function as an oncogene is int-2, a member of the fibroblast growth factor family. Int-2 is sometimes activated in mouse mammary carcinomas by MMTV insertional mutagenesis.
Growth Factor Receptors
Some viral oncogenes are altered versions of normal growth factor receptors that possess intrinsic tyrosine kinase activity. Receptor tyrosine kinases, as these growth factor receptors are collectively known, have a characteristic protein structure consisting of three principal domains: (1) the extracellular ligand-binding domain, (2) the transmembrane domain, and (3) the intracellular tyrosine kinase catalytic domain). Growth factor receptors are molecular machines that transmit information in a unidirectional fashion across the cell membrane. The binding of a growth factor to the extracellular ligandbinding domain of the receptor results in the activation of the intracellular tyrosine kinase catalytic domain. The recruitment and phosphorylation of specific cytoplasmic proteins by the activated receptor then trigger a series of biochemical events generally leading to cell division.
Because of the role of growth factor receptors in the regulation of normal cell growth, it is not surprising that these receptors constitute an important class of protooncogenes. Examples include erb B, erb B-2, fms, kit, met, ret, ros, and trk. Mutation or abnormal expression of growth factor receptors can convert them into oncogenes. For example, deletion of the ligand-binding domain of erb B (the epidermal growth factor receptor) is thought to result in constitutive activation of the receptor in the absence of ligand binding. Point mutation in the tyrosine kinase domain or of the extracellular domain and deletion of intracellular regulatory domains can also result in the constitutive activation of receptor tyrosine kinases. Increased expression through gene amplification and abnormal expression in the wrong cell type are additional mechanisms through which growth factor receptors may be involved in neoplasia. The identification and study of altered growth factor receptors in experimental models of neoplasia have contributed much to our understanding of the normal regulation of cell proliferation.
Signal Transducers
Mitogenic signals are transmitted from growth factor receptors on the cell surface to the cell nucleus through a series of complex interlocking pathways collectively referred to as the signal transduction cascade. This relay of information is accomplished in part by the stepwise phosphorylation of interacting proteins in the cytosol. Signal transduction also involves guanine nucleotide-binding proteins and second messengers such as the adenylate cyclase system. The first retroviral oncogene discovered, src, was subsequently shown to be involved in signal transduction.
Many protooncogenes are members of signal transduction pathways. These consist of two main groups: nonreceptor protein kinases and guanosine triphosphate (GTP)-binding proteins. The nonreceptor protein kinases are subclassified into tyrosine kinases (eg, abl, lck, and src) and serine/threonine kinases (eg, raf-1, mos, and pim-1). GTP-binding proteins with intrinsic GTPase activity are subdivided into monomeric and heterotrimeric groups. Monomeric GTP-binding proteins are members of the important ras family of protooncogenes that includes H-ras, K-ras, and N-ras. Heterotrimeric GTP-binding proteins (G proteins) implicated as protooncogenes currently include gsp and gip. Signal transducers are often converted to oncogenes by mutations that lead to their unregulated activity, which in turn leads to uncontrolled cellular proliferation.
Transcription Factors
Transcription factors are nuclear proteins that regulate the expression of target genes or gene families. Transcriptional regulation is mediated by protein binding to specific DNA sequences or DNA structural motifs, usually located upstream of the target gene. Transcription factors often belong to multigene families that share common DNA-binding domains such as zinc fingers. The mechanism of action of transcription factors also involves binding to other proteins, sometimes in heterodimeric complexes with specific partners. Transcription factors are the final link in the signal transduction pathway that converts extracellular signals into modulated changes in gene expression.
Many protooncogenes are transcription factors that were discovered through their retroviral homologs. Examples include erb A, ets, fos, jun, myb, and c-myc. Together, fos and jun form the AP-1 transcription factor, which positively regulates a number of target genes whose expression leads to cell division. Erb A is the receptor for the T3 thyroid hormone, triiodothyronine. Protooncogenes that function as transcription factors are often activated by chromosomal translocations in hematologic and solid neoplasms. In certain types of sarcomas, chromosomal translocations cause the formation of fusion proteins involving the association of EWS gene with various partners and resulting in an aberrant tumor-associated transcriptional activity. Interestingly, a role of the adenovirus E1A gene in promoting the formation of fusion transcript fli1/ews in normal human fibroblasts was recently reported. An important example of a protooncogene with a transcriptional activity in human hematologic tumors is the c-myc gene, which helps to control the expression of genes leading to cell proliferation. As will be discussed later in this chapter, the cmyc gene is frequently activated by chromosomal translocations in human leukemia and lymphoma.
Programmed Cell Death Regulation
Normal tissues exhibit a regulated balance between cell proliferation and cell death. Programmed cell death is an important component in the processes of normal embryogenesis and organ development. A distinctive type of programmed cell death, called apoptosis, has been described for mature tissues. This process is characterized morphologically by blebbing of the plasma membrane, volume contraction, condensation of the cell nucleus, and cleavage of genomic DNA by endogenous nucleases into nucleosome-sized fragments. Apoptosis can be triggered in mature cells by external stimuli such as steroids and radiation exposure. Studies of cancer cells have shown that both uncontrolled cell proliferation and failure to undergo programmed cell death can contribute to neoplasia and insensitivity to anticancer treatments.
The only protooncogene thus far shown to regulate programmed cell death is bcl-2. Bcl-2 was discovered by the study of chromosomal translocations in human lymphoma. Experimental studies show that bcl-2 activation inhibits programmed cell death in lymphoid cell populations. The dominant mode of action of activated bcl-2 classifies it as an oncogene. The bcl-2 gene encodes a protein localized to the inner mitochondrial membrane, endoplasmic reticulum, and nuclear membrane. The mechanism of action of the bcl-2 protein has not been fully elucidated, but studies indicate that it functions in part as an antioxidant that inhibits lipid peroxidation of cell membranes. The normal function of bcl-2 requires interaction with other proteins, such as bax, also thought to be involved in the regulation of programmed cell death. It is unlikely that bcl-2 is the only apoptosis gene involved in neoplasia although additional protooncogenes await identification.
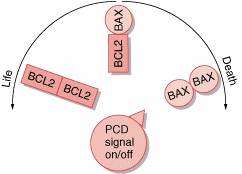
Mechanisms of oncogene activation
The activation of oncogenes involves genetic changes to cellular protooncogenes. The consequence of these genetic alterations is to confer a growth advantage to the cell. Three genetic mechanisms activate oncogenes in human neoplasms: (1) mutation, (2) gene amplification, and (3) chromosome rearrangements. These mechanisms result in either an alteration of protooncogene structure or an increase in protooncogene expression. Because neoplasia is a multistep process, more than one of these mechanisms often contribute to the genesis of human tumors by altering a number of cancer-associated genes. Full expression of the neoplastic phenotype, including the capacity for metastasis, usually involves a combination of protooncogene activation and tumor suppressor gene loss or inactivation.
Mutation
Mutations activate protooncogenes through structural alterations in their encoded proteins. These alterations, which usually involve critical protein regulatory regions, often lead to the uncontrolled, continuous activity of the mutated protein. Various types of mutations, such as base substitutions, deletions, and insertions, are capable of activating protooncogenes. Retroviral oncogenes, for example, often have deletions that contribute to their activation. Examples include deletions in the aminoterminal ligand-binding domains of the erb B, kit, ros, met, and trk oncogenes. In human tumors, however, most characterized oncogene mutations are base substitutions (point mutations) that change a single amino acid within the protein.
Point mutations are frequently detected in the ras family of protooncogenes (K-ras, H-ras, and N-ras). It has been estimated that as many as 15% to 20% of unselected human tumors may contain a ras mutation. Mutations in K-ras predominate in carcinomas. Studies have found K-ras mutations in about 30% of lung adenocarcinomas, 50% of colon carcinomas, and 90% of carcinomas of the pancreas. N-ras mutations are preferentially found in hematologic malignancies, with up to a 25% incidence in acute myeloid leukemias and myelodysplastic syndromes. The majority of thyroid carcinomas have been found to have ras mutations distributed among K-ras, H-ras, and N-ras, without preference for a single ras family member but showing an association with the follicular type of differentiated thyroid carcinomas. The majority of ras mutations involve codon 12 of the gene, with a smaller number involving other regions such as codons 13 or 61. Ras mutations in human tumors have been linked to carcinogen exposure. The consequence of ras mutations is the constitutive activation of the signal-transducing function of the ras protein.
Another significant example of activating point mutations is represented by those affecting the ret protooncogene in multiple endocrine neoplasia type 2A syndrome (MEN2A).
Germline point mutations affecting one of the cysteines located in the juxtamembrane domain of the ret receptor have been found to confer an oncogenic potential to the latter as a consequence of the ligand-independent activation of the tyrosine kinase activity of the receptor. Experimental evidences have pointed out that these mutations involving cysteine residues promote ret homodimerization via the formation of intermolecular disulfide bonding, most likely as a result of an unpaired number of cysteine residues.
Gene Amplification
Gene amplification refers to the expansion in copy number of a gene within the genome of a cell. Gene amplification was first discovered as a mechanism by which some tumor cell lines can acquire resistance to growth-inhibiting drugs. The process of gene amplification occurs through redundant replication of genomic DNA, often giving rise to karyotypic abnormalities called double-minute chromosomes (DMs) and homogeneous staining regions (HSRs). DMs are characteristic minichromosome structures without centromeres. HSRs are segments of chromosomes that lack the normal alternating pattern of light- and dark-staining bands. Both DMs and HSRs represent large regions of amplified genomic DNA containing up to several hundred copies of a gene. Amplification leads to the increased expression of genes, which in turn can confer a selective advantage for cell growth.
The frequent observation of DMs and HSRs in human tumors suggested that the amplification of specific protooncogenes may be a common occurrence in neoplasia. Studies then demonstrated that three protooncogene families-myc, erb B, and ras-are amplified in a significant number of human tumors. About 20% to 30% of breast and ovarian cancers show c-myc amplification, and an approximately equal frequency of c-myc amplification is found in some types of squamous cell carcinomas. N-myc was discovered as a new member of the myc protooncogene family through its amplification in neuroblastomas. Amplification of N-myc correlates strongly with advanced tumor stage in neuroblastoma, suggesting a role for this gene in tumor progression. L-myc was discovered through its amplification in small-cell carcinoma of the lung, a neuroendocrine-derived tumor. Amplification of erb B, the epidermal growth factor receptor, is found in up to 50% of glioblastomas and in 10% to 20% of squamous carcinomas of the head and neck. Approximately 15% to 30% of breast and ovarian cancers have amplification of the erbB-2 (HER-2/neu) gene. In breast cancer, erbB-2 amplification correlates with advanced stage and poor prognosis. Members of the ras gene family, including K-ras and N-ras, are sporadically amplified in various carcinomas.
Chromosomal Rearrangements
Recurring chromosomal rearrangements are often detected in hematologic malignancies as well as in some solid tumors. These rearrangements consist mainly of chromosomal translocations and, less frequently, chromosomal inversions. Chromosomal rearrangements can lead to hematologic malignancy via two different mechanisms: (1) the transcriptional activation of protooncogenes or (2) the creation of fusion genes. Transcriptional activation, sometimes referred to as gene activation, results from chromosomal rearrangements that move a proto-oncogene close to an immunoglobulin or T-cell receptor gene. Transcription of the protooncogene then falls under control of regulatory elements from the immunoglobulin or T-cell receptor locus. This circumstance causes deregulation of protooncogene expression, which can then lead to neoplastic transformation of the cell.
Fusion genes can be created by chromosomal rearrangements when the chromosomal breakpoints fall within the loci of two different genes. The resultant juxtaposition of segments from two different genes gives rise to a composite structure consisting of the head of one gene and the tail of another. Fusion genes encode chimeric proteins with transforming activity. In general, both genes involved in the fusion contribute to the transforming potential of the chimeric oncoprotein. Mistakes in the physiologic rearrangement of immunoglobulin or T-cell receptor genes are thought to give rise to many of the recurring chromosomal rearrangements found in hematologic malignancy. In some cases, the same protooncogene is involved in several different translocations (ie, c-myc, ews, and ret).
Gene Activation
The t(8;14)(q24;q32) translocation, found in about 85% of cases of Burkitt lymphoma, is a well-characterized example of the transcriptional activation of a proto-oncogene. This chromosomal rearrangement places the c-myc gene, located at chromosome band 8q24, under control of regulatory elements from the immunoglobulin heavy chain locus located at 14q32. The resulting transcriptional activation of c-myc, which encodes a nuclear protein involved in the regulation of cell proliferation, plays a critical role in the development of Burkitt lymphoma. The c-myc gene is also activated in some cases of Burkitt lymphoma by translocations involving immunoglobulin light-chain genes. These are t(2;8)(p12;q24), involving the κ locus located at 2p12, and t(8;22)(q24;q11), involving the κ locus at 22q11. Although the position of the chromosomal breakpoints relative to the c-myc gene may vary considerably in individual cases of Burkitt lymphoma, the consequence of the translocations is the same: deregulation of c-myc expression, leading to uncontrolled cellular proliferation.
In some cases of T cell acute lymphoblastic leukemia (T-ALL), the c-myc gene is activated by the t(8;14)(q24;q11) translocation. In these cases, transcription of c-myc is placed under the control of regulatory elements within the T-cell receptor α locus located at 14q11. In addition to c-myc, several protooncogenes that encode nuclear proteins are activated by various chromosomal translocations in T-ALL involving the T-cell receptor α or β locus. These include HOX11, TAL1, TAL2, and RBTN1/Tgt1. The proteins encoded by these genes are thought to function as transcription factors through DNA-binding and protein-protein interactions. Overexpression or inappropriate expression of these proteins in T cells is thought to inhibit T-cell differentiation and lead to uncontrolled cellular proliferation.
A number of other protooncogenes are also activated by chromosomal translocations in leukemia and lymphoma. In most follicular lymphomas and some large cell lymphomas, the bcl-2 gene (located at 18q21) is activated as a consequence of t(14;18)(q32;q21) translocations. Overexpression of the bcl-2 protein inhibits apoptosis, leading to an imbalance between lymphocyte proliferation and programmed cell death. Mantle cell lymphomas are characterized by the t(11;14)(q13;q32) translocation, which activates the cyclin d1 (bcl-1) gene located at 11q13. Cyclin D1 is a G1 cyclin involved in the normal regulation of the cell cycle. In some cases of T cell chronic lymphocytic leukemia and prolymphocytic leukemia, the tcl-1 gene at 14q32.1 is activated by inversion or translocation involving chromosome 14. The tcl-1 gene product is a small cytoplasmic protein whose function is not yet known.
Gene Fusion
The first example of gene fusion was discovered through the cloning of the breakpoint of the Philadelphia chromosome in chronic myelogenous leukemia (CML). The t(9;22)(q34;q11) translocation in CML fuses the c-abl gene, normally located at 9q34, with the bcr gene at 22q11. The bcr/abl fusion, created on the der(22) chromosome, encodes a chimeric protein of 210 kDa, with increased tyrosine kinase activity and abnormal cellular localization.111 The precise mechanism by which the bcr/abl fusion protein contributes to the expansion of the neoplastic myeloid clone is not yet known. The t(9;22) translocation is also found in up to 20% of cases of acute lymphoblastic leukemia (ALL). In these cases, the breakpoint in the bcr gene differs somewhat from that found in CML, resulting in a 185 kDa bcr/abl fusion protein. It is unclear at this time why the slightly smaller bcr/abl fusion protein leads to such a large difference in neoplastic phenotype.
In addition to c-abl, two other genes encoding tyrosine kinases are involved in distinct gene fusion events in hematologic malignancy. The t(2;5)(p23;q35) translocation in anaplastic large cell lymphomas fuses the NPM gene (5q35) with the ALK gene (2p23). ALK encodes a membranespanning tyrosine kinase similar to members of the insulin growth factor receptor family. The NPM protein is a nucleolar phosphoprotein involved in ribosome assembly. The NPM/ALK fusion creates a chimeric oncoprotein in which the ALK tyrosine kinase activity may be constitutively activated. The t(5;12)(q33;p13) translocation, characterized in a case of chronic myelomonocytic leukemia, fuses the tel gene (12p13) with the tyrosine kinase domain of the PDGF receptor b gene (PDGFR-b at 5q33). The tel gene is thought to encode a nuclear DNA-binding protein similar to those of the ets family of protooncogenes.
Gene fusions sometimes lead to the formation of chimeric transcription factors. The t(1;19)(q23;p13) translocation, found in childhood pre-B-cell ALL, fuses the E2A transcription factor gene (19p13) with the PBX1 homeodomain gene (1q23). The E2A/PBX1 fusion protein consists of the amino-terminal transactivation domain of the E2A protein and the DNA-binding homeodomain of the PBX1 protein. The t(15;17)(q22;q21) translocation in acute promyelocytic leukemia (PML) fuses the PML gene (15q22) with the RARA gene at 17q21. The PML protein contains a zinc-binding domain called a RING finger that may be involved in protein-protein interactions. RARA encodes the retinoic acid alpha-receptor protein, a member of the nuclear steroid/thyroid hormone receptor superfamily. Although retinoic acid binding is retained in the fusion protein, the PML/RARA fusion protein may confer altered DNA-binding specificity to the RARA ligand complex. Leukemia patients with the PML/RARA gene fusion respond well to retinoid treatment. In these cases, treatment with all-trans retinoic acid induces differentiation of PML cells.
The ALL1 gene, located at chromosome band 11q23, is involved in approximately 5% to 10% of acute leukemia cases overall in children and adults. These include cases of ALL, acute myeloid leukemia, and leukemias of mixed cell lineage. Among leukemia genes, ALL1 (also called MLL and HRX) is unique because it participates in fusions with a large number of different partner genes on the various chromosomes. Over 20 different reciprocal translocations involving the ALL1 gene at 11q23 have been reported, the most common of which are those involving chromosomes 4, 6, 9, and 19. In approximately 5% of cases of acute leukemia in adults, the ALL1 gene is fused with a portion of itself. This special type of gene fusion is called self-fusion. Self-fusion of the ALL1 gene, which is thought to occur through a somatic recombination mechanism, is found in high incidence in acute leukemias with trisomy 11 as a sole cytogenetic abnormality. The ALL1 gene encodes a large protein with DNA-binding motifs, a transactivation domain, and a region with homology to the Drosophila trithorax protein (a regulator of homeotic gene expression). The various partners in ALL1 fusions encode a diverse group of proteins, some of which appear to be nuclear proteins with DNA-binding motifs. The ALL1 fusion protein consists of the aminoterminus of ALL1 and the carboxyl terminus of one of a variety of fusion partners. It appears that the critical feature in all ALL1 fusions, including self-fusion, is the uncoupling of the ALL1 amino-terminal domains from the remainder of the ALL1 protein.
Solid tumors, especially sarcomas, sometimes have consistent chromosomal translocations that correlate with specific histologic types of tumors. In general, translocations in solid tumors result in gene fusions that encode chimeric oncoproteins. Studies thus far indicate that in sarcomas, the majority of genes fused by translocations encode transcription factors. In myxoid liposarcomas, the t(12;16)(q13;p11) fuses the FUS (TLS) gene at 16p11 with the CHOP gene at 12q13. The FUS protein contains a transactivation domain that is contributed to the FUS/CHOP fusion protein. The CHOP protein, which is a dominant inhibitor of transcription, contributes a protein-binding domain and a presumptive DNA-binding domain to the fusion. Despite knowledge of these structural features, the mechanism of action of the FUS/CHOP oncoprotein is not yet known. In Ewing sarcoma, the t(11;22)(q24;q12) fuses the EWS gene at 22q12 with the FLI1 gene at 11q24. Like FUS, the EWS protein contains three glycine-rich segments and an RNA-binding domain. The FLI1 protein contains an ets-like DNA-binding domain. The EWS/FLI1 fusion protein combines a transactivation domain from EWS with the DNA-binding domain of FLI1. In alveolar rhabdomyosarcoma, the t(2;13)(q35;q14) fuses the PAX3 gene at 2q35 with the FKHR gene at 13q14. The PAX3 protein, a transcription factor that activates genes involved in development, is a paired-box homeodomain protein with two distinct DNA-binding domains. The FKHR protein encodes a conserved DNA-binding motif (the forkhead domain) similar to that first identified in the Drosophila forkhead homeotic gene. The PAX3/FKHR fusion protein is a chimeric transcription factor containing the PAX3 DNA-binding domains, a truncated forkhead domain, and the carboxy-terminal FKHR regions.
In DP, an infiltrating skin tumor, both a reciprocal translocation t(17;22)(q22;q13) and supernumerary ring chromosomes derived from the t(17;22) have been described. Although early successful studies in this field have been performed with lymphomas and leukemia, as we have discussed before, the first chromosomal abnormality in solid tumors to be characterized at the molecular level as a fusion protein was an inversion of chromosome 10 found in papillary thyroid carcinomas. In this tumor, two main recurrent structural changes have been described, including inv(10) (q112.2; q21.2), as the more frequent alteration, and a t(10;17)(q11.2;q23). These two abnormalities represent the cytogenetic mechanisms which activate the protooncogene ret on chromosome 10, forming the oncogenes RET/ptc1 and RET/ptc2, respectively. Alterations of chromosome 1 in the same tumor type have then been associated to the activation of NTRK1 (chromosome 1), an NGF receptor which, like RET, forms chimeric fusion oncogenic proteins in papillary thyroid carcinomas. A comparative analysis of the oncogenes originated from the activation of these two tyrosine kinase receptors has allowed the identification and characterization of common cytogenetic and molecular mechanisms of their activation. In all cases, chromosomal rearrangements fuse the tK portion of the two receptors to the 5′ end of different genes that, due to their general effect, have been designated as activating genes. In the majority of cases, the latter belong to the same chromosome where the related receptor is located, 10 for RET and 1 for NTRK1. Furthermore, although functionally different, the various activating genes share the following three properties: (1) they are ubiquitously expressed; (2) they display domains demonstrated or predicted to be able to form dimers or multimers; (3) they translocate the tK-receptor-associated enzymatic activity from the membrane to the cytoplasm.
These characteristics can explain the mechanism(s) of oncogenic activation of ret and NTRK1 protooncogenes. In fact, following the fusion of their tK domain to activating gene, several things happen: (1) ret and NTRK1, whose tissue-specific expression is restricted to subsets of neural cells, become expressed in the epithelial thyroid cells; (2) their dimerization triggers a constitutive, ligand-independent transautophosphorylation of the cytoplasmic domains and as a consequence, the latter can recruit SH2 and SH3 containing cytoplasmic effector proteins, such as Shc and Grb2 or phospholipase C (PLCγ), thus inducing a constitutive mitogenic pathway; (3) the relocalization in the cytoplasm of ret and NTRK1 enzymatic activity could allow their interaction with unusual substrates, perhaps modifying their functional properties.
In conclusion, in PTCs, the oncogenic activation of ret and NTRK1 protooncogenes following chromosomal rearrangements occurring in breakpoint cluster regions of both protooncogenes could be defined as an ectopic, constitutive, and topologically abnormal expression of their associated enzymatic (tK) activity.
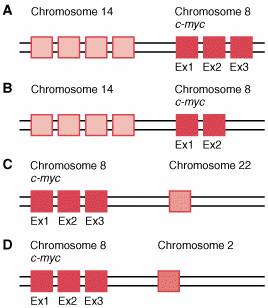
C-myc translocations found in Burkitt lymphoma.
A, t(8;14)(q24;q32) translocation involving the locus of immunoglobulin heavy-chain gene located at 14q32. B, t(8;14)(q24;q32) translocation where only 2 exons (Ex) of c-myc are translocated under regulatory elements from the immunoglobulin heavy-chain locus located at 14q32. C, t(8;22)(q24;q11) translocation involving the l locus of immunoglobulin light-chain gene at 22q11. D, t(2;8)(p12;q24) translocation involving the κ locus of immunoglobulin light-chain gene located at 2p12.