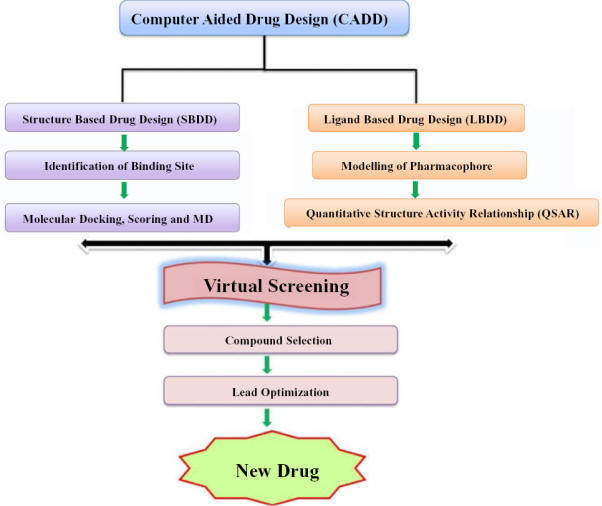
computer-aided drug design (CADD)
The
application of computers and computational methods in the field of drug design
and discovery process is referred to as computer-aided drug design (CADD). It is
beneficial in the hit-to-lead discovery, lead optimization which drastically
reduced the time and cost factor involved in the new drug discovery process.
Modeling three-dimensional structures of ligand and protein, simulation,
prediction of binding interactions and energy is a challenging job in the field
of drug design. Most of the molecular modeling methods are based on molecular
mechanics or quantum mechanics, although both the methods generate equations for
calculating total energy of the system but differ from each other in some
fundamental aspects.
Broadly, CADD methods are classified
into two categories and are structure-based drug design (SBDD) and ligand-based
drug design (LBDD).
Structure-based drug design (SBDD)
A
high-resolution protein structure or a prepared homology model of the protein is
a vital need of structure-based designing. Protein structural information like
binding sites, cavities, secondary binding sites, etc., is highly useful for the
discovery of small-molecule binding agents which can modulate biological
activity. This information is required for ascertaining the molecular
interactions of the ligand within the binding cavity. The main target of SBDD is
to design and discover ligand molecules with high binding affinity and of
complementary features. SBDD includes docking, molecular dynamics, and
pharmacophore modeling.
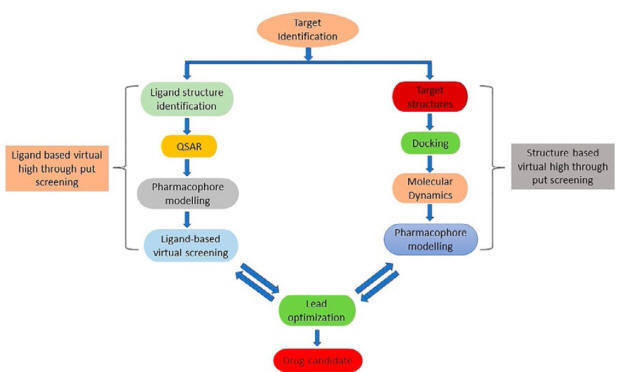
Target Identification
Drug target identification and its
validation is the initial step of the drug discovery process. It is a
macromolecule that has an established function in the pathophysiology of a
disease. Four major drug targets are found in organisms, i.e. proteins,
including receptors and enzymes, nucleic acids (DNA and RNA), carbohydrates, and
lipid. The majority of drugs available in the market are addressed to proteins
as a target. However, due to the decoding of several genomes of pathogens,
nucleic acids could gain big importance as drug targets in the future. The
selection of potential drug targets from thousands of candidate macromolecules
is a challenging task. In the post-genomic era, genomics and proteomics
approaches are the most important tools for target identification. Besides, advances in high-throughput omics technologies
generated a huge amount of data for host–pathogen interaction. These available data are also integrated and
analyzed by the scientific community through
network and systems biology approaches to accelerate the process of target
identification in drug
discovery program.
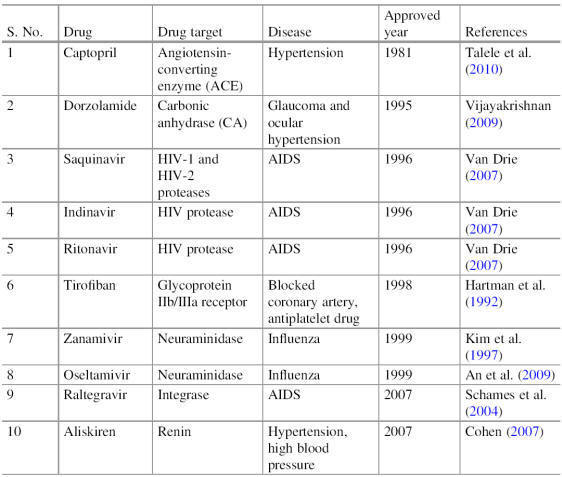
Drug Targets
The term drug target describes the native biomolecule in the human body whose
function can be modulated by a drug molecule, which may have a therapeutic
effect against the disease or some adverse effect. Mostly these drug targets are
biological targets in nature. Various protein drug
targets are currently utilized by available drugs, most of them belong to one of
four major drug target protein classes (Table), in some cases, nucleic acids are
also utilized by drugs as a target.
Details of frequently used drug
target protein classes


http://hit2.badd-cao.net/
Databases for therapeutic target
information

Drug Target
Identification
After identifying the biological nature and origin of a disease, identification
of potential drug targets is the
first step in the discovery of a
drug. Drug target identification follows the hypothesis
that the most promising targets are tightly linked to the disease of interest,
and have an established function in the underlying pathology, which can be
observed with high frequency in the disease-associated population. By definition,
it is not necessary for potential drug targets to be involved in the
disease-causing process, or responsible for a disease, but they must be disease
modifying.
Currently, various strategies are in practice for drug target identification,
which is either based on experimental approaches or computational approaches.
Experimental approaches are mainly based on comparative genomics (expression profiling)
and supplemented with the phenotype and genetic association analysis. Mostly,
all experimental approaches provide reliable results, and theoretically,
theyshould be the
first-choice methods for target
identifications. Even though
experimental approaches are more precise, they are suffering from some practical
limitations, i.e., relatively high costs and intensive scientific
labor required for experimental profiling of the full target space (>20,000
proteins, nucleic acid) of chemical compounds and they often end with few drug
targets in hand. Due to all these limitations, mostly scientists and
pharmaceutical companies utilize the computational methods for
first-line
research and then use the experimental approaches for further validation and
other purposes.
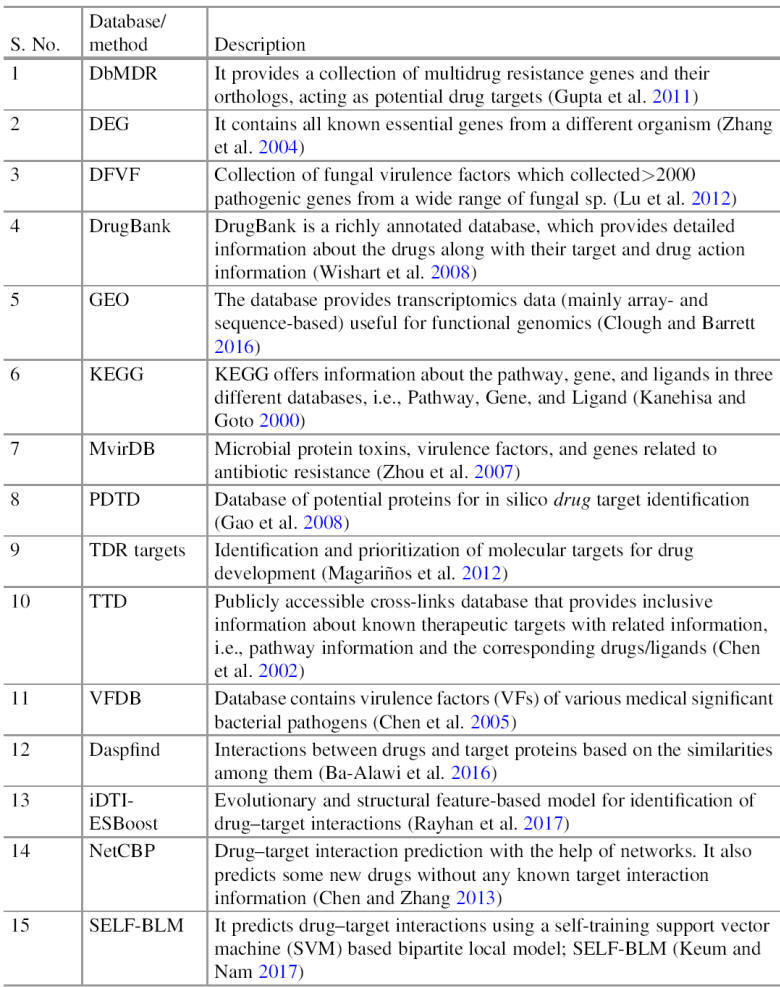
Computational Approaches for Drug Target Identification
The development of bioinformatics has come up with various
bioinformatics resources, including the database, algorithm, and software, which
push the CADD in every aspect of the drug designing process (Table). One of the
most important contributions is computational drug target identification, as discussed earlier that identification of the drug target is a very crucial and most decisive step
of the drug designing process. In this regard, for the last one and half
decades, various scientific studies carried
out with the aim of drug target identification with the help
of bioinformatics resources and proposed various approaches for drug target
identifications. These
approaches easily handle and deal with a huge amount of genomics,
transcriptomics, and proteomics data, and also process it efficiently, and at the end provide potential drug targets in a short
period at a low cost.
Currently, several computational approaches are available which
utilized different molecular information, i.e., gene and genome sequence,
molecular interaction information and protein 3D structure. Most of these
approaches are interlinked. Still, based on their concept, they have broadly
classified into two types:
(1) homology-based approaches and
(2) network-based approaches.
The major features which are checked for drug target prediction are
listed in Table.
Important features utilized in
drug target identifications

Homology-Based
Approaches
Homology-based approaches utilize sequence similarities among genes and
proteins, further based on predicted homology, it takes the decision just like
decision tree analysis. Mostly these methods consider the various level of
homology test, which follows top-down direction. Each level of homology test
scale down the data, starting from complete genes or
proteome, and step by step either eliminate those which
fitted in
“inappropriate”
or select only those
which
fitted in
“appropriate.”
Homology-based
approaches always ended with countable potential drug targets (Fig.), and
because of their scale down nature, these approaches are also known as
subtractive (genomic or proteomic) approaches.
The term
“inappropriate”
and
“appropriate”
are conditional, and they are
tested on various biological conditions that play a decisive role in target
selection. The following are the major conditional tests that help to decide the
further consideration of molecules for drug target identification.
Bioinformatics resources for
drug target identification and CADD
Schematic diagram of the
standard
flowchart for drug target identification
using homology-based approach

Human Homologs
It is assumed that humans have various genes, and few of them are playing an
indispensable biological role, considered as housekeeping genes. The use of
human housekeeping genes or homologs of human housekeeping genes as a drug
target can create lethal conditions and result in the death of human patients.
To avoid such accidental use of the housekeeping gene as well as some important
pathway-related gene as a drug target gene of the microbial pathogen are
generally compared against the human, and those genes which show significant similarities with human
housekeeping or crucial genes will be considered as
“inappropriate”
and mostly eliminate from rest
of the process.
Human-Microbiome
Homologs
The human body, especially, the gut has a lot of microbes that are already
listed by the human microbiome project. Most of these microbes are involved in
the biological process, which is beneficial for humans and thus
considered beneficial microbes. Use of homologs
from these beneficial microbes as a drug target
can harm these bacteria, which can affect the related biological process in the
human host, i.e., digestion, respiration process, etc., because of the above
said reason, human-microbiome homologs are considered as
“inappropriate”
and eliminated from the further
process.
Essentiality
Identification
of drug targets against the microbial pathogen assumes that the essentiality of
the target protein for pathogen-microbes is one of the advantageous and
“appropriate”
features. Without the function
of essential proteins, microbial pathogen will not able to survive. Various
essential genes and proteins are identified by experimental approaches
and enlisted in various databases. The database of essential genes (DEG) is one
of the most active databases providing a collection of essential genes and
protein sequences. Based on the above concept, those pathogenic genes/proteins
which show homology with essential genes/proteins are considered as
“appropriate”
and include for the further
process.
Virulence Factor
Homologs
Those proteins whose role in virulence and pathogenicity is reported through the
experiment are considered as virulence factors. Various such proteins are
available, especially for microbes, and their molecular information is stored in
various databases, i.e., virulence factor database (VFDB) and database of fungal
virulence factors (DFVF). Genes/proteins of the pathogens that show homology
with these virulence factors can be considered as
“appropriate”
and utilized as a potential drug
target.
Drug Target
Homologs
Information about known and explored drug/therapeutic targets is available,
i.e., therapeutic target database (TTD). Homology mining with TTD is in
practice, and those candidate molecules which show significant homology with these known
targets are considered as
“appropriate”
and included for further
exploration.
Cellular Location
The cellular location of the target protein is one of the very important
features and plays a crucial role in target selection. In a homology-based
approach, sequence-based gene ontology (GO) and annotation are in practice to
look at the sub-cellular location along with the cellular component, biological
process, and molecular function. Generally, those targets whose access is easy
are preferable over others.
Role in the
Biological Pathway
Biological pathways are responsible for the synthesis or metabolism of various
bio-products. Few of these pathways are very important and unique, and they are
solely responsible for their processes and products. The blockage of these
pathways creates a scarcity of their products and
finally reduces the chance of
survival of the pathogen. Various pathway databases are available to conduct
such checks. Current literature shows that the KEGG pathway is one of the
richest and preferable pathway databases utilized for this purpose. Those
pathways which are unique for pathogen are considered as appropriate pathways,
and gene/proteins involved in them were considered for the further process. In
contrarily those pathways which are also shared by human/host and their
gene/proteins are
“inappropriate”
and excluded from further
consideration.
It has been observed that homology-based approaches are very fast and almost
cover the entire target space, and it only needs sequence information as input.
Available reviews suggest that uses of homology-based approaches are very
common for microbial disease and generally restricted
with them only. Their use for other types of infection or disease is not in
common practice.
Subtractive approach for drug
target identification
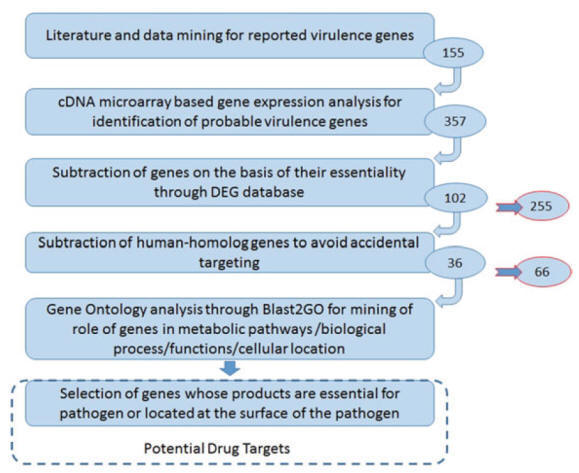
Case
Study: Subtractive Approach for Drug Target Identification
The subtractive approach is one of the very famous approaches that
have been utilized for target identification against
various pathogens. In 2011 Katara et al. presented a subtractive approach
exploiting the knowledge of global gene expression along with sequence
comparisons to predict the potential drug targets in
Vibrio cholerae, cholera causing
bacterial pathogen, efficiently. Their
analysis was based on the available knowledge of 155 experimentally proved
virulence genes (seed information) (Fig.). For target identification, they utilized co-expression based gene mining and
multilevel subtractive approach. At the end, they reported 36 gene products as a
drug target, to check the reliability of the predicted targets they also
performed gene ontology through Blast2GO. They observed these targets for their
involvement in a crucial biological process and their cellular location. They
found all these 36 gene products as reliable targets and conclude them as
potential drug targets.
Network-Based Approaches
It examines the effects of drugs in the context of molecular
networks (i.e., protein–
protein
interactions, gene networks, transcriptional regulatory networks, metabolic
networks, and biochemical reaction networks). In molecular network models,
molecules refer as nodes, and each edge corresponds to an interaction between
two molecules, based on the direction and importance of interaction between
nodes, sometimes edges also mention the direction and weight (Fig.). Drug target
identification through the
network is based on the fact that networks have many important nodes that are
vulnerable and can be targeted in many ways. Most of the time, these nodes are
very crucial, and sometimes essential for the whole network structure,
inhibition of such nodes can reduce their efficiency and damage of
these nodes can shut down the complete network. Network inhibition process
follows one of the following two models:
(1) partial inhibitions: Partial knockout of the interactions of
the target nodes, and
(2) complete inhibition: all interactions around a given target
node are eliminated.
In the drug designing process, these target nodes can be considered
as potential drug targets. Various molecular networks (Table), including
protein-interaction networks, regulatory, metabolic, and signaling networks
individually or in integrated form can be subjected to a similar analysis.
Various components of a standard
network
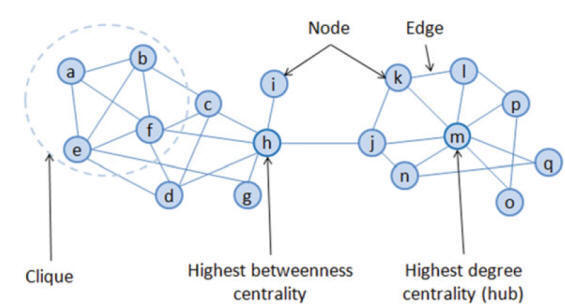
Clique :
A clique
is a fully connected
graph /
group with same characteristics.
Types of the biological network
for drug target identification

Centrality Based Drug Target
Network centrality can be used as a potential tool for
network-based target identification. Network
centrality can prioritize proteins based on the network centrality measures
(i.e., degree, closeness betweenness). It can be used to characterize the
importance of proteins in the biological system.
Hubs
as Target
Real-world networks almost show a scale-free degree distribution,
which means that in these networks, some nodes have a tremendous number of
connections to other nodes (high degree), whereas most nodes have just a few.
Here, nodes with a great number of connections than average called hubs. It
assumes that the functionality of such scale-free networks heavily depends on
these hubs, and if these hubs are selectively targeted, the information transfer
through networks gets hindered and results in the collapse of the network.
Betweenness Centrality Based Target
Hubs are the centers of local network topology, thus only provide
the local picture of the network. Betweenness centrality is another approach
that can be used to explain network centre, unlike, hub it provides central
elements of the network in the global topology, thus, provide a global picture
of network connections. Conceptually, betweenness is the number of times a node
is in the shortest paths between two other nodes (Fig.), thus higher the
betweenness means more importance of the node in quick network communication.
Such higher betweenness centrality nodes can be utilized as a potential target
against drugs.
Mesoscopic Centrality Based Target
Considering the advantage of both local and global centers of
network topology for drug target identifications, the third
class of centrality called mesoscopic centrality has also been reported.
Mesoscopic centrality is neither fully based on local
information (such as
hubs) nor global information (such as betweenness centrality) on network
structure. It mainly considers long-range connections between high degree nodes,
which make a profound effect on small-world networks.
Weight-Based Drug Target
Recently, the weighted-directed network is also reported for drug
target identification studies. The
weighted-directed network is closer to the real, cellular scenario, where PPIs
are characterized by their affinity and dominance
(link weight) as well as direction (e.g., in form of signaling), as mentioned in
Fig. It has been assumed that the deletion of the links with the highest
weighted centralities is often more disturbing to network behavior than the
removal of the most central links in the similar un-weighted network topology.
Utilization of the complex structural information of real-world
networks to measure the centrality is not an easy task, and it requires more
sophisticated methods to overcome these challenges. Bioinformatics provides
various tools to support network construction, visualization, and network-based
analysis, i.e., weight, centrality, interaction directions (Table).
Molecular network with a
different type of connectivity between nodes (A) undirected (B) directed (C) weighted, and (D) weighted directed

Tools supporting molecular
network analysis for drug target identification

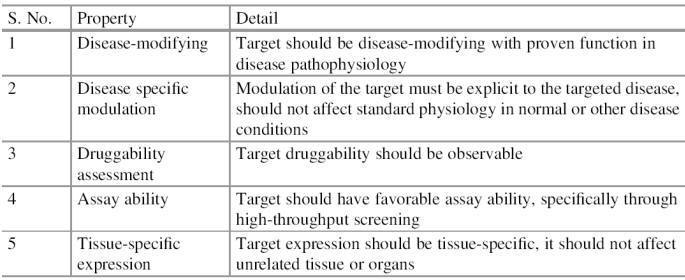
Properties of an Ideal Drug Target
Identification of potential
drug targets is not the last step. Nowadays, through various computational
approaches, a huge number of probable targets are reported against different
diseases and are available in databases and literature. It is not a good idea to
recommend them directly for testing, its recommendation that
first, we check them for an ideal property (Table), and then for
druggability. Only those targets which fulfill most of them are
considered as an ideal drug target and recommended them for further validation
and testing.
Important properties to assess
the ideal drug targets
Chemical databases

Chemical Information
Chemical databases are considered as a powerful tool in drug design
and discovery. Possible requirements-based searches in the database can
find molecules with desired biological activity that might be an
appropriate candidate for further analysis. Some important resource databases
for chemical information are summarized in Table.
In the early stages of drug discovery,
researchers are testing thousands of natural products or plant extracts, small
molecules, and looking for a potential molecule to develop as a drug. If they
get a potential hit candidate, they move it to the next stage of hit-to-lead
optimization. Therefore, the
first stage of
a significant drug
finding
project is
“hit identification.”
In this
practice, hits or small chemical compounds are identified, which
bind to the protein and modify its task. So, hits ideally show some degree of
specificity and
potency against the target.
A compound which shows activity
against the desired target when tested in a suitable assay is named as hit. A ch
emical entity with structural and reproducible activity data. It involves a
screening of a wide range of small molecules in an
in
vitro
assay. The molecules with
in
vitro
potency (IC50 value) less than 20
mM are considered as hits. The hit validation process ensures the
activity of the molecules
in
vivo
and generates potential lead compounds. Hits free from potential
toxicity are called as drug-like compounds.
(A)
Application of computer-aided drug design (CADD) techniques in various drug
discovery stages,
(B)
application of CADD-based virtual screening for lead identification.
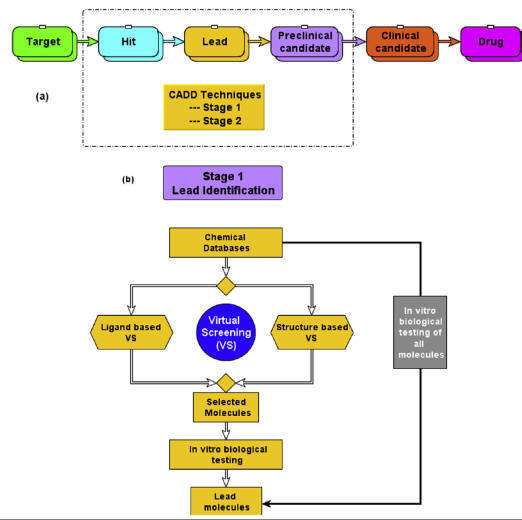
Lead discovery and development
Lead discovery and development
involves the identification of a synthetic or natural chemical
molecule/peptide/antibody that specifically
and efficiently binds to a drug target and thereby modulates
its biological function. Lead molecules (also referred to as hit) could be
considered as a prototype, from which the drug molecules are developed. The
initial step in lead discovery is to identify a starting molecule that shows
reasonable biological activity toward the target protein. Both experimental and
computational methods are widely used for lead identification
and optimization as well. Some of the extensively used experimental methods that
are being used for hit identification include high-throughput screening (HTS),
combinatorial library screening, knowledge-based screening, fragment-based
screening, etc. Alternatively, computational methods commonly known as CADD
techniques such as virtual screening (VS) methods have emerged as powerful
techniques for hit identification. In VS experiments, a molecular library
consisting of millions of chemical molecules is screened computationally (in
silico) in a short time and the compounds which are predicted as active/positive
are then subjected for further biological testing, whereas the
filtered
inactive/negative compounds are skipped from biological testing. This VS
strategy for lead identification significantly
reduces the cost and workload as compared to HTS screening.
Combinatorial chemistry
Combinatorial chemistry comprises the methodology in which a large number of
chemical libraries were prepared using a single process and tested for the
biological activity. This technology works on the principle of production of a
large number of chemical compounds of given methods for performing biological
actions. The advantage of the techniques is the reduction in time and money
which augments the drug development process. The combinatorial chemistry
approaches are appreciated and widely accepted by the pharmaceutical sector for
drug designing and screening. The combinatorial chemistry approaches are
generally divided into two broad classes:
Unbiased library
This is also well known as “random library,” in which the libraries were
designed through the synthetic approach concepts without concern about the
molecular target. The synthesized library compounds having diversity in the
chemical structure were used for the hit identification library for the concern
target.
Biased library
The
biased library approaches the building blocks, scaffold were utilized for the
generation of different library types. This technique is limited to a particular
building block and is focused on a selective target for biological activity.
Solid-phase synthesis
The
solid-phase synthesis involves the synthesis of compounds using functionalized
solid supports. The solid supports used are polymeric beads and insoluble
resin-type materials. In this, the linker groups are attached to the resins
beads solid support, and then starting materials or reactants were passed
through the solid support to form the intermediates. The library of compounds
was synthesized using split and mix techniques such as biological oligomers and
polymeric peptide synthesis. Finally, the product was obtained after the
detachment from the linker solid support from washing and purification
techniques.
Advantages
·
Solid support provides easy purification.
·
Excess reagent can be used, providing more yield.
Disadvantages
·
Limited numbers of reactions
·
More reaction time required
·
More reagents requirement
·
Monitoring is difficult
·
Expensive
Solution-phase synthesis
The
solution-phase synthesis is another alternative method for the synthesis of a
number of lead molecules. This method has one of the main disadvantages
regarding purification techniques to obtain the desired product from the
reaction mixture. Therefore, to overcome these difficulties further
technological advancement is done in purification methods to obtain the desired
product with good quantity and yields.
The
techniques used for purification involve the following:
·
Polymer-supported reagents and scavengers
·
Liquid-liquid extraction techniques
·
Chromatography techniques.
Despite the purification problem in the solution-phase synthesis, it is
considered an important tool in combinatorial chemistry for library synthesis.
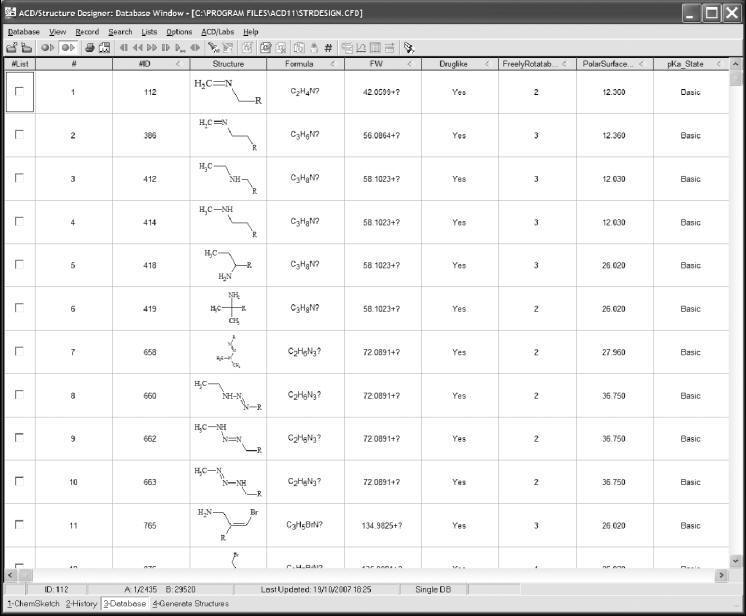
Compound library design
Compound library design usually refers to the generation of a list of structures
to be synthesized through combinatorial synthesis. There are a number of
approaches that drug designers can utilize in performing this task. Designers
may get fairly deep into the experimental plate design, or they may design a
library as a collection of compounds that researchers would like to test,
without regard for the synthesis route. There are a numberof software tools for
aiding with library design, which have a correspondingly diverse range of
functionality.
Targeted libraries versus diverse libraries
One issue to be considered is whether the library is to be narrowly focused (a
targeted library) or very diverse. In the earlier stages of a design project,
diverse libraries will often be used in order to explore a wide range of
chemistries. Later, strongly focused groups of compounds (possibly differing
only by a single functional group) will be synthesized and tested. In general,
it is easier to design a narrowly targeted library. This is done to explore
possible derivatives of a known structure, usually for the purpose of increasing
activity. It is typically done by first selecting a backbone structure (often a
fused ring system), and then selecting synthons to be used to create derivatives
of that structure. The designer can identify a point on the molecule to be
altered, and select a bioisosteric group of functional groups to put at that
point. The term “synthon” refers to a functional group to be added at a
particular point. Synthons are typically molecular fragments with unfilled
valence positions, not reagents to be used in a synthetic reaction.
Focused libraries are also easier to synthesize. It is often possible to use the
same chemical reaction for all of the compounds, with just one reagent
substituted. Designing diverse chemical libraries tends to be a more difficult
task.
Researchers must contend with some rather difficult questions of chemistry:
·
How diverse should the library be?
·
Are there any two compounds that are too similar?
·
Are there gaps in the chemistries represented, where an
additional compound with those specifications should be included?
·
Does the library span the space of known chemistries, or known
drug-like chemistries?
Often, a diverse library is created by selecting compounds from a list of
compounds already synthesized and available either in inventory or from
commercial sources. This is more cost-effective than trying to synthesize a very
diverse set of compounds for each drug design project. Late in the drug design
process, focused libraries are made for the purpose of improving
bioavailability, half-life in the bloodstream, and toxicity. Figure shows an
example of a library design tool specifically for this purpose.
The ACD/Structure
Designer program makes modifications specifically for the purpose of improving
the pharmacokinetic properties of an active lead.
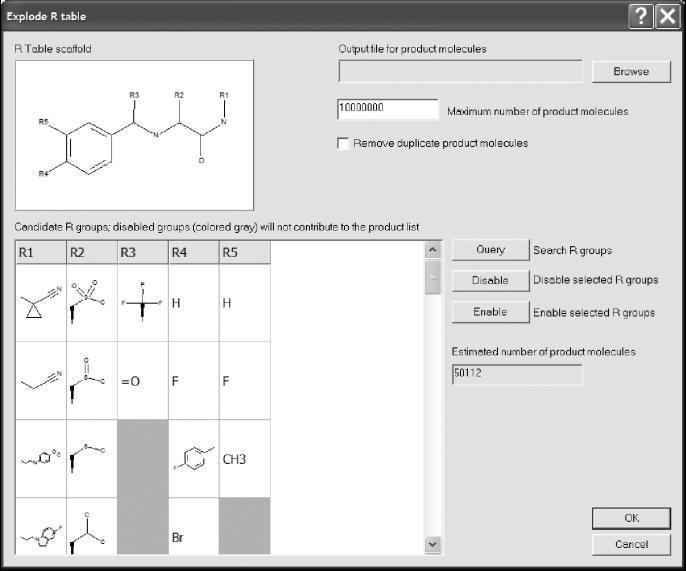
From fragments versus from reactions
Most library design tools work by allowing the user to suggest the functional
group lists; the tool can then generate structures for the compounds that would
be created from those lists. When examining the “nuts and bolts” of how library
design tools work, there are two different approaches for defining the reagent
lists: the fragment approach and the reaction approach. In the fragment
approach, backbones and side chains are defined with an open valence location
defined as a dummy atom, instead of a hydrogen. This fragment is called a
synthon. The program can then generate a list of product compounds by connecting
the functional groups in the list to the backbones, knowing that the dummy atom
is the point of connection. The advantages of this approach are that the
researcher can stay focused on the resulting set of molecules and that there are
no ambiguities in how the researcher intends to connect the pieces together. The
disadvantage is that the design process is rather disconnected from the
synthesis process. Some programs can generate fragment lists automatically from
a list of compounds, and some require that every fragment be edited by hand to
define the connection points. Figure shows an example of a fragment-based
library design program.
Class Pharmer from Simulations Plus is a library design tool that uses a
fragment based approach.
The alternative is to work with a piece of software that allows the researcher
to define a chemical reaction and the list of reagents. The advantage of this is
that it is closer to the synthesis route, so the results do not often come up
with compounds that cannot practically be synthesized with the intended
reaction. This can bring to light ambiguities in the synthesis when there are
multiple functional groups, thus making it possible to create several different
products from the chosen reactants. Regardless of which type of library creation
tool is used, the majority of the work at the library design stage is in the
process of selecting the backbones and synthons.
Non-enumerative techniques
Most library design tools are enumerative techniques. This means that the
functional group lists are used to generate structures, in the computer’s
memory, for every compound that can be synthesized in the library. The entire
list of structures can then be feed into various types of prediction software.
Enumerative techniques workwell for designing small to moderate-size compound
libraries, containing up to thousands or tens of thousands of compounds.
Non-enumerative techniques are useful for manipulating very large library
designs. The backbone and functional group lists are generated, just as they are
in an enumerative algorithm. However, in a non-enumerative algorithm, the
structures for each individual compound are never generated. Group additivity
methods are used to compute properties for the library, such as minimum,
maximum, and estimated average molecular properties. However, only numbers that
describe the entire library are generated in this way-values for individual
compounds are never computed.
The advantage of non-enumerative methods is that they can be used to explore
large, diverse chemical spaces representing billions of compounds, in a way that
would not be practical, or even possible, using other techniques. This is a
different type of software to work with. Users must get used to thinking in
terms of a large theoretical chemical space, instead of being able to see
individual molecular structures.
Virtual screening
The
virtual screening method is based on a comparative analysis between different
leads (candidates for new drug candidates), using computational resources,
generally based on the corollary that the drug’s action is directly related to
its affinity for a biological target. In this context, docking is the most used
technique, which consists of the interaction between a molecule and a biological
target, calculating the interaction energy between them. The calculations
generally employ molecular dynamics, making it
possible to obtain interaction free energies and, from there, the inhibition
constants (an experimental parameter that can be verified). There are online
resources that enable virtual screening, often comparing interaction energies
with biological activity data.
The use of artificial intelligence techniques has been
fundamental in the systematization of these studies. Virtual tissue and organ
models are an elegant solution to this approach, being able to anticipate even
physiological and neural phenomena.
Virtual screening techniques
Drug discovery programs have been considered as challenging and slow processes
with a high failure rate. To reduce the burden of cost and time, pressure has
been mounting on researchers to identify and separate unsuitable drug candidates
in early drug discovery phases.
Although
high-throughput screening equipped with combinatorial synthesis has been a front
step of “hit to lead” identification, reducing a substantial amount of time,
still it suffers from drawbacks like consuming valuable resources and time.
In the past two decades, virtual screening has been rapidly developed to make
the drug discovery process more fast, cheap, and reliable. Virtual screening is
an in silico method which uses various scoring and ranking functions to screen a
large number of databases or yet-to-be synthesized chemical structures against a
specific biological target.
The concept of virtual screening has been developed from the
pioneer works of Kuntz et al. and Desjarlais et al.
However, Horvath coined the term
“virtual screening” in his research paper based on trypanothione reductase
inhibitors.
This led to the evolution of a new
concept in the field of computational drug design for searching new bioactive
agents from a database of compounds. These agents are screened based on the
structural parameters, predicted to be complementary to a specific molecular
target or enzyme. Advancements in the field of computer hardware and algorithms
led to the progress and widespread use of virtual screening as a computational
method in the drug discovery process. The significance of virtual screening
assisted with other in silico tools can be realized from the fact that more than
50 drug candidates have got green signal to proceed through clinical trials, and
some of them also got approved for clinical use.
Chemical space
It is the hypothetical space containing all the possible chemical structures,
which probably ranges from 1018 to 10180 molecules.
Analyzing the infinite chemical space
for searching bioactive compounds is like finding a needle in the haystack. As
the drug candidates belong to various diverse sources (synthetic, natural,
marine, peptides, microorganism, etc.), instead of entire chemical
space-specific regions a relevant biological target has to be searched. Hence,
putting biological activity as a filter enables virtual screening to find the
specific biologically active regions of chemical space.
Several bioactive compounds failed to become successful drug
candidates because of unfavorable physicochemical properties, which adversely
affect their absorption, distribution, metabolism, and elimination (ADME). This
led to the evolution of drug-likeness and ADME parameters, which facilitated the
virtual screening procedure and its success rate in finding the active medicinal
space.
Database
An essential part of virtual screening includes preparation of compounds
database, where compounds can be stored in 2D (SMILES) or 3D chemical structure
formats (MDL SD, Sybyl mol2, CML, PDB, XYZ). Another open-source format
developed by IUPAC is International Chemical Identifier (InChI) which can encode
chemical structures and is able to identify various protomeric and tautomeric
states.
These structures are usually
annotated with other information like molecular weight, synthetic source, amount
available, stereochemistry, tautomer, conformers, and protonation state. These
structural data along with various physicochemical as well as biological
properties help in screening a database by removing the undesirable compounds,
which in turn enriches the database with desirable compounds. Researchers showed
keen interest in annotated compounds databases, which have information regarding
both chemical structure and its possible biological activity. Therefore, it has
gradually succeeded over traditional compound databases containing information
on chemical structures only.
Drug molecules collected from various sources have been broken into
individual fragments using the retrosynthetic principle and again combined in
every possible way to create a library of virtual compounds. Virtual
combinatorial libraries have a tremendous impact in extending the diverse range
of chemical space, which are now available for screening. A few examples of databases (proteins, nucleic acids, complexes as
well as ligands) available in the public domain for free as well as commercial
use are as follows: AntiBase, BindingDB, BraMMT (Brazilian Malaria Molecular
Targets), ChEMBL, ChemSpider, CMNPD, COlleCtion of Open NatUral producTs
(COCONUT), DrugBank, DrugSpaceX, EDULISS, eMolecules, GOSTAR, MCDB, MDDR (MDL
Drug Data Reports), MMsINC, OOMT (Our Own Molecular Targets), PubChem, and ZINC.
Identification of a bioactive compound against a specific biological target
having minimal adverse effects is the primary goal of a drug discovery program.
But the presence of promiscuous compounds, frequent hits, and screening
artifacts can overwhelm the actual active compounds, which poses a great
challenge before the researchers.
Pharmacological promiscuous compounds, which act on multiple biological targets,
often come out as successful hits in virtual screening, but later on they are
found to be nondrug-like. They have a noncompetitive mechanism and poor
selectivity as well as a structure-activity relationship.
When certain compounds interfere with the assay method and give a false-positive
result, they are called artifacts in virtual screening. Various filters are
applied to identify and remove such type of nuisance compounds for improving the
efficiency of virtual screening.
Classification
Depending on the knowledge of biological target structure, virtual screening
techniques can be broadly categorized into two types: structure-/target-based
virtual screening and ligand-based virtual screening.
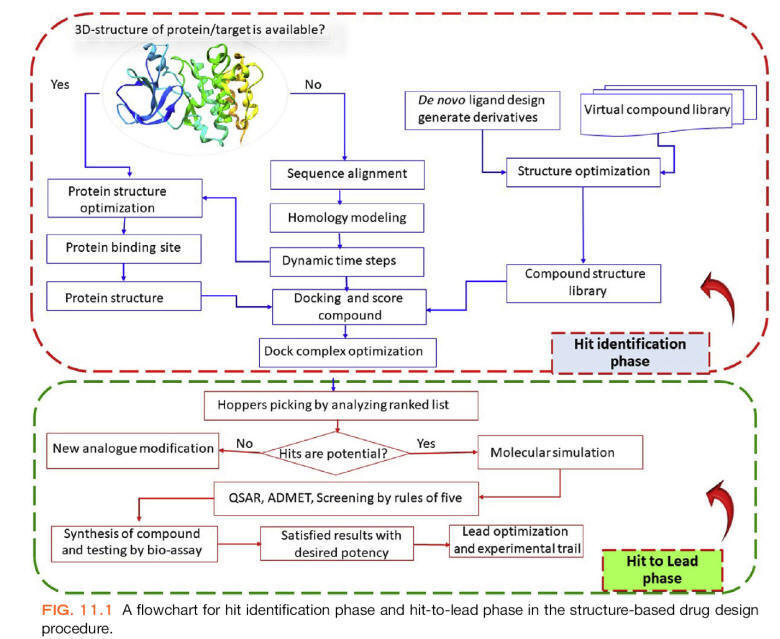
Structure- or target-based virtual screening
It involves the ranking of ligands as per their affinity with the biological
target, as evinced by the nature of interactions during the formation of the
ligand-receptor complex.
The ligands may be screened or categorized based on their affinity toward
different biological targets.
Knowledge about 3D structure of receptors is essential, which are developed
either by X-ray crystallography, NMR spectroscopy, electron microscopy or by
homology modeling.
The protein structure must be checked for structural disorder or missing
residues which can be rectified. Identification of a binding site within the
protein structure is another prerequisite. A binding site may be associated with
metal ions or water molecules which play an important role in ligand binding,
and this information is essential during setting up a virtual screening. A
number of binding site detection algorithms are being used by various
computational tools for the identification of binding sites inside the protein
structure.
Docking and receptor-based
pharmacophore modeling are the two elementary methods for carrying out
structure-based virtual screening.
Docking
Since the development of the molecular docking technique, it has been proposed
to be used as a filter in virtual screening.
Docking is an in-silico method which predicts the interactions and
probable binding conformation of ligand molecules inside the binding cavity of
receptor structure. Prediction of interactions, less computational time, and
cost make docking a preferred method for executing virtual screening. A typical
docking-based virtual screening consists of four steps: ligand
setup/preparation, protein setup/preparation, docking, and postdocking analysis.
The large database of compounds should be downsized before docking for removing
false positives and unsuitable structures by applying filters like 2D or 3D
pharmacophoric features, druglikeness properties, chemical reactivity, etc.
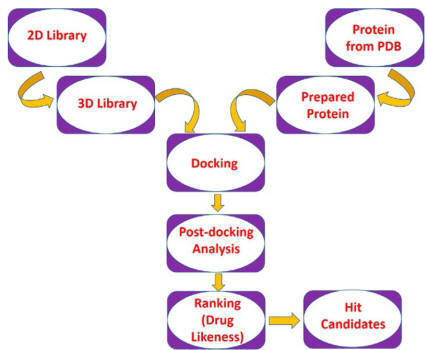
Application of constraints in virtual screening
Various constraints which are applied in virtual screening can be divided into
three classes. Covalent interaction-based constraints can filter out ligands
having specific covalent interactions with the receptor.
Conformational space-based constraints can screen ligands
occupying a specific region in the conformational space of the binding site.
Pharmacophoric or scaffold-based
constraints may be applied as either predocking filters
to filter out unsuitable structures,
or as postdocking filters
to select similar binding poses, which satisfies the pharmacophoric criteria.
Alternatively, shape and similarity-based constraints and motif-based
constraints may also be utilized in virtual screening to ensure a similar
binding manner of different ligands. A flowchart of docking-based virtual
screening is presented in
Fig.
Demerits
Docking methods could not perform simulations with sufficient receptor
flexibility or on protein with an induced-fit mechanism. Simulations of ligand
and receptor in a polar medium, illustrating the effect of metal ions and
assigning correct protonation state to the atoms, could not be achieved by
docking.
Receptor-based pharmacophore modelling
Structure- or receptor-based pharmacophore modeling can be applied in virtual
screening for getting new leads from a database of compounds. This method can
describe the molecular interactions within the binding site and emerge as an
alternative method of virtual screening by overcoming the barriers observed with
the docking method. Various pharmacophoric features are mapped onto the ligand
structure to derive structural information and interactions responsible for
making a compound bioactive. It helps to retrieve structures with diverse
bioisosteric scaffolds, which is difficult to explore by systematic
derivatization of known compounds. Several pharmacophoric models can be aligned
together to create a common pharmacophore hypothesis or model.
This hypothesis can be used as a
filter for screening the database to find hits with novel scaffolds, e.g., FLAP,
GBPM, GRID, LigandScout, MOE (Molecular Operating Environment;
http://www.chemcomp.com/),
and Unity (Tripos;
http://www.tripos.com/).
Ligand-based virtual screening
This method is used when the protein structure is not known. The structure of
known active and inactive compounds is used as templates, based on which
algorithms search for new compounds having structural similarity with the
templates. Ligand-based virtual screening can be performed with the help of
three methods: similarity search, ligand-based pharmacophore modeling, and
machine learning method.
Similarity search
The structure of a single active compound is the minimum information we need for
similarity searching. The selection of appropriate descriptors is very important
in carrying out similarity search-based screening. A compound with known
activity is taken as a template or reference, based on which a library of
compounds can be screened and ranked accordingly. It can be performed by using
either a molecular alignment algorithm or molecular descriptors/fingerprint
algorithm.
Ligand-based pharmacophore modelling
When receptor structure is not known, ligand-based pharmacophore modeling can be
used for virtual screening. A single/group of compounds(s), with known activity
against a specific target, can be analyzed to identify different chemical
features from its structure. Different conformations are generated for each
molecule and then aligned together to map the corresponding features. However,
two different approaches can be used for the generation of ligand-based
pharmacophore models:
(i) A database with predefined conformations for each ligand can be used as a
filter to speed up the screening, which needs a huge storage facility for
handling a huge number of conformations; and
(ii) a single conformation of a known active compound can be used for generating
different
conformations, followed by aligning these conformations with the database
structures to generate models, which can be used as a filter to screen the
database. Although it does not need much storage facility, it is very slow.
Catalyst, Disco, and GASP are some of the programs that utilize the ligand-based
pharmacophore method.
Machine learning method
It is an application of artificial intelligence to create a model, built on a
group of experimentally determined actives and inactives.
It can predict the activity of an
unknown compound against a specific target and also can distinguish active
compounds from the inactive ones. Regression models can be built by using
training set compounds which correlate activity with the structural information.
The machine learning method also utilizes the information of inactive compounds
to harvest structure-activity relationship among the dataset compounds. These
generated models may be used as a filter in screening large databases.
Variouspredicted ADME and other properties can be utilized along with the
machine learning method to downsize the hit list. These are of two types:
unsupervised and supervised.
Unsupervised methods
These methods utilize the descriptor information to correlate the biological
activity with the dataset structures. It helps in identifying a specific region
of the dataset containing predominantly active or inactive compounds. Since few
parameters are used to build robust models, overfitting does not happen with
this method, e.g., principal component analysis (PCA), K-means clustering, and
self-organizing map.
Supervised methods
A group of compounds or a subset of the total dataset molecules with known
actives and inactives are selected to form a training set to build a model. The
remaining dataset compounds (test set) are used to examine the predictive
capacity of the model, known as cross-validation, which is essential to avoid
overfitting. Several models are built by taking different training and test set
compounds, out of which a single best cross-validated model is chosen, e.g.,
decision tree (recursive partitioning), K-nearest neighbor, artificial neural
networks, and support vector machines.
Successful applications of virtual screening
A ligand-based virtual screening of approximately 718,000 commercially available
compounds was carried out based on three known glucocorticoid receptor
antagonists as query structures. A 3D molecular similarity-based filter and
clustering technique was utilized to downsize the database, which followed by
lead identification found a compound with good activity (Ki=16 nM). Further optimization led to
the discovery of CORT118335, phase II clinical candidate for management of
nonalcoholic steatohepatitis and schizophrenia.
Virtual screening was carried out on the AZ corporate
database of about 1 million compounds based on 10 known fibrinolysis inhibitors
as query molecules. A 3D electrostatic and shape-based similarity approach was
utilized to obtain an active compound 4-PIOL, which on optimization led to the
clinical trial candidate AZD6564 for treatment hemorrhage.
Liang et al. have successfully screened the covalent natural products database
using herb-based mapping to identify the active compounds baicalein and baicalin
showing PLK-1 inhibitory activity.
Burggraaff et al. have successfully carried out a statistical and
structure based virtual screening for the discovery of several RET kinase
inhibitors. Rollinger et al. discovered two novel acetylcholinesterase
inhibitors (scopoline and scopoletin) by structure-based pharmacophore screening
of 110,000 natural products database by using protein structure and a known
inhibitor.
Advanced computational resources are
being implemented in virtual screening to search lead compounds and to assist
hit finding procedure by preselecting compounds for biological evaluation.
Although the positive hits obtained from the virtual screening must undergo
experimental screening, it saves time and valuable resources which get wasted on
the synthesis and activity of random compounds. Undoubtedly, virtual screening
makes the drug discovery process fast, efficient, and more economic.
Virtual high-throughput screening (vHTS)
In the pharmaceutical sector, costs of developing new drugs molecules are high
despite increased spending on rising technologies. To cut costs linked with the
development of new drugs, computational techniques have been established. Among
these computational methods is the virtual high-throughput screening (vHTS),
handout tools to quest novel drugs with the capability to bind a specific
molecular target in the process of drug discovery. In the drug discovery
approach, vHTS utilizes computational algorithms to detect novel bioactive
compounds. It rises the hit rate of novel compounds as it practices more
extensive target exploration as equated to old combination chemistry and
high-throughput screening, which elucidates the molecular characteristics
responsible for therapeutic action aiding the prediction of probable derivatives
with enhanced activity. It is a computer-assisted technique in the drug
development process that can cut back time and costs, also lowering the omission
rate by prioritizing molecules for more tentative examination.
A typical procedure in the virtual
high-throughput screening is shown in
Fig.
Tools for virtual high-throughput screening (vHTS)
Some of the available tools for vHTS are Octopus, PyRx Virtual Screening Tool,
and Raccoon2.
Octopus
It
is a virtually automated road-mapping organization tool for performing vHTS. It
combined user-friendly molecular docking interface AutoDock Vina, PyMOL, and
MGLTools to execute vHTS. Octopus can process molecular docking data for an
unrestricted sum of ligands and molecular targets, which cannot be executed in
other platforms like PyRx and Raccoon2. During vHTS, it can efficiently operate
numerous molecules versus a group of targets. Additionally, it comprises
molecular targets databank for malaria, dengue, and cancer. Conveniently, it can
also lessen the quantity of biological assays required to discover a
pharmacological mechanism. It is predominantly restricted by the time required
to draw the ligands’ structures and the choice of preferred targets. Apart from
a visual inspection carried out by computational chemists for the intermolecular
interactions of the molecules and target, the complete procedure can run
automatically.
PyRx
It
is open-source software for virtual screening of small molecules with a
convenient intuitive interface that can operate on Mac OS, Windows, and Linux
operating systems. In PyRx, to initiate the virtual screening, structures of the
target molecules are required as input files. The input files can be download
from a number of freely accessible Web sites such as DrugBank, PubChem, and
Protein Data Bank. By default, the integrated AutoDock Vina docking interface
outputs the 10 best binding modes for every course of docking. The handlers can
also export virtual screening outcomes as SDF files as well as comma-separated
values (CSV), which further enhances the analysis and filtering of virtual
screening results suitable for third-party packages.
Raccoon2
It
is a preparatory and analyzing platform of a graphical interface for virtual
screenings that utilizes AutoDock Vina as the default docking program. Raccoon2
is a better-developed platform with more flexibility and robustness than the
former version Raccoon as the code has been revised from scratch and designed to
augment new features such as the investigational characteristics of Fox,
allowing simplified examination of outcome, outlining information, and sharing
structures among different associated laboratories. Raccoon2 can run only in
Linux computational platform with PBS and SGE schedulers. Some of the features
of Raccoon2 are automated downloading feature with capable preprocessing of
experimental outcomes and filtering of the results by properties (energy, ligand
efficiency) and interactions.
Techniques for virtual high-throughput screening (vHTS)
There are basically two approaches to this topic: ligand- and structure based
vHTS.
Ligand-based vHTS
When the structure of the target is unknown, the measured activities for some
known compounds can be used to construct a pharmacophore model. Ligand based
approaches apply ligand details grounded on resemblance or diversity to earlier
identified active ligands to forecast activity. Ligands displaying parallel
activity to an active ligand are likely to be more potent than random ligands
with few tweaks in the key structural characteristics, such as placement of
hydrogen bond and hydrophobic groups; the latter helps in selecting a template
with the most capable contenders from the library.
Ligand-based vHTS can be categorized into the following classes
Fingerprint-based methods:
There is anticipation in the molecules to possess similar characteristics when
there is a structural resemblance. Databases are comprised of compounds with
similar structures possessing unknown biological activity and may comprise
compounds with some desired activity.
Generalized pharmacophoric method
Pharmacophore is a structural feature accountable for compounds’ activity at a
receptor site. In this method, common pharmacophore layout is identified for a
set of known active molecules and the layout is afterward used for the search of
a 3D substructure. The software assists to find the spatial arrangement in atoms
and functional groups, which match this component with the interrogate molecule.
Machine learning approaches
In the machine learning approaches, logic based directions are carried out to
define the properties of the substructures associated with biological activity
on a training set of data comprising identified active and inactive molecules.
It delivers insights into activity.
Structure-based vHTS
Structure-based vHTS depends on the 3D structural information of the target
protein procured by different techniques such as crystallographic,
spectroscopic, and bioinformatics to estimate the reaction energy of the tested
compound. Structure-based vHTS involves the finding of a possible binding site
of ligand on the target molecules; subsequently, docking of ligands to a target
protein with a scoring purpose to evaluate the affinity of a ligand will bind to
the target protein. It has been developed to a basis of computational biology
and medicinal chemistry, enriching the understanding of the biological target
and the chemistry behind the ligand-protein interactions.
The important stages in structure-based vHTS are:
·
Preparation of library for ligand.
·
Preparation of target protein.
·
Confirmation of most promising binding position.
·
Grading of ligand-protein docked complexes.
In this technique, for a complete set of virtual compounds database binding
modes on the target protein are determined by a docking program.
Applications of virtual high-throughput screening (vHTS)
It
is used for three major purposes:
(1)
To refine a large set of compounds to a relatively smaller set of prevised
compounds, for experimentally testing later;
(2)
To enhance the affinity of lead compounds by improving the pharmacokinetic
assets such as absorption, distribution, metabolism, excretion, and toxicity
(ADMET); and
(3)
To design novel active candidates by mounting start molecules with different
functional groups or organizing fragments into a novel chemical entity.
Approved antiviral drugs such as saquinavir, ritonavir, and indinavir are some
of the drugs discovered by computer-aided drug design.
Pharmacophore
A
pharmacophore is an active part of the molecules that states the necessary
features which are responsible for the biological interaction and therapeutic
activity. The pharmacophore of any molecules determined active functional groups
of ligands that interact with the receptor target which promotes the biological
inhibition or activation effects of the protein target. The molecular
interaction with the receptor of the ligand depends on the pharmacophore
features involving polar, nonpolar, aromatic, ring, and charges. These features
show different types of interactions with the amino acid residues of the
receptor, such as hydrogen bonds with the polar, hydrophobic interactions, and
aromatic interactions. The pharmacophore feature in the morphine compound is
tertiary alkylamine, and this structure permits the interaction with opioid
receptors and responses the biological action through an activated signal
cascade that results in the analgesia and sedation-type property.
Pharmacophore modeling and similarity search
Through pharmacophore screening, it is possible to identify compounds containing
different scaffolds, but with a similar3Darrangement of key interacting
functional groups, onto which binding site information can be incorporated.
Structure-based pharmacophore modeling
As
per IUPAC, pharmacophore is very well defined as “A pharmacophore is the
ensemble of steric and electronic features that is necessary to ensure the
optimal supramolecular interactions with a specific biological target structure
and to trigger (or to block) its biological response.”
Various pharmacophoric features include
hydrogen bond donor/acceptor, charged center (positive or negative), hydrophobic
and aromatic region, metal-binding region, distance, angle, and dihedral angles.
When these pharmacophoric features along with their 3D distributions are
assigned within the binding site, it can reveal the structural information and
interactions responsible for making a compound bioactive.
This method can also retrieve structures with diverse bioisosteric scaffolds,
which is difficult to explore by systematic derivatization of known compounds.
Analysis of various favorable interactions between ligand and the receptor
binding site can be classified under hydrogen bonds (donors/acceptors),
electrostatic charge centers (positive/negative), and hydrophobic contacts to
generate pharmacophore models. Several pharmacophoric models can be aligned
together to produce a common pharmacophore hypothesis or model.
Pharmacophore modeling has been utilized for searching databases,
virtual screening, fragment designing, and scaffold hopping and for forecasting
bioactivity of hypothetical compounds.
Ligand-based pharmacophore modeling
In
case of nonavailability of protein structure, ligand-based pharmacophore
modeling can be employed. It has two essential steps:
(1)
pharmacophoric features in training set compounds are
analyzed;
(2) aligning all the active conformations of ligands in training set based on
chemical features or molecular field descriptors.
Ligand-based pharmacophore models are of two types:
Qualitative models: A set of
active ligands (no explicit biological activity data required) of diverse
structures are used to generate a common feature based pharmacophoric
hypothesis.
Quantitative models:
A set of known active compounds (activity data expressed in
Ki
or IC50) are utilized to create
QSAR-based predictive pharmacophoric models.
Examples: Catalyst includes two alternative
algorithms like HypoGen and HipHop for building pharmacophore-based models.
HypoGen assigns a certain weighting factor to each of the chemical features of
the ligand responsible for bioactivity and constitutes a pharmacophore model. In
this way, several pharmacophore hypotheses can be prepared and ranked as per
ability to correlate the bioactivity. HipHop explores the surface accessibility
of the active ligands suitable for interactions with the receptor to determine
their absolute coordinates. Pharmacophore models are prepared based on the
chemical features assigned to the absolute coordinates in different
conformations. A number of pharmacophore hypotheses can be generated and ranked
based on their ability to explain bioactivity.
Disco program adopts a different approach of breaking the pharmacophore into
ligand points (hydrogen bonds, charged centers, hydrophobic region) and binding
pocket interaction sites (complementary regions within the receptor and is
mapped by the coordinates of heavy atoms of ligand). Like catalyst, here also a
set of predefined conformations limit the ability to explore the entire
conformational space of the ligand.
GASP program utilizes genetic algorithm to search the conformational space to
generate different models. A ligand with a minimum number of common chemical
features is considered as a reference or template structure. All other compounds
(present in the training set) are aligned to this template to evaluate the
fitness of a specific pharmacophore model based on similarity, overlaid
features, and volume integral of the overlay. Unlike Catalyst and Disco, here
overall shape along with any steric clashes between the ligands is taken into
consideration during the generation of the final model.
Quantitative structure-activity relationship (QSAR)
QSAR may be defined as the application of statistical approaches, namely
regression and classification methods, on the pursuing of quantitative
relationships between the biological activity of a set of congener compounds and
their structural, topological, electronic, electrotopological, steric, and
physicalchemical properties (among others). This knowledge field assumes that
the behavior of a substance in the biological environment depends on its
structural characteristics, which affect its overall properties.
Historical background
While the discovery of new therapeutic agents was restricted to the empiricism
of isolation from natural sources until the mid-eighteenth century, in 1868
Crum-Brown and Fraser,2
working with derivatives of morphine, strychnine, and atropine in guinea pigs,
postulated that the physiological action of a drug was a function of its
chemical structure. In 1893, Richet
established that the toxicity of a series of alcohols, ethers, and
ketones was inversely proportional to their solubility in water (which was
ultimately related to lipophilia, although this concept was not yet fully
established). Overton and Meyer,
in 1899, postulated that the narcotic activity of some derivatives was a
function of the partition coefficient between chloroform and water, which
augmented alongside the set of compounds, suffering a decrease for very
lipophilic compounds. This behavior was associated with the fact that compounds
with a very high affinity for lipids would not be able to reach with ease the
site of action, being retained In the guinea pig’s adipose tissues. Ferguson’s
works,
in 1939, established the equivalence
relationship between the chemical potential of exobiophase and the chemical
potential of endobiophase, which allows making inferences about the intensity of
the drug’s action in the internal phase by measuring it in the external phase.
Quantitative structure-activity relationship (QSAR)
From a set of similar molecules, physicochemical descriptors (electronic,
hydrophobic, steric), topological, among others, are obtained. A multivariate
regression analysis is performed between these descriptors and biological
activity, resulting in an equation that represents a model of the system
investigated. The 3D-QSAR approach uses the three-dimensional structures of
compounds, properly aligned, on which a probe (for example, a positive carbon)
is placed around a grid of points, calculating the steric and electrostatic
energy. Each value of these energies becomes a physicochemical descriptor in the
model that uses biological activity data as a dependent variable. The points of
interaction favorable or unfavorable to biological activity are converted into a
three dimensional map showing regions favored by large groups and regions
favored by small groups, and regions favored by positive groups and those
favored by these groups.
QSAR modeling
The
conventional approach of structure-activity relationship (SAR) in drug design
and discovery has brought many successes but not without a great degree of luck.
QSAR method correlates various quantifiable physicochemical
properties with biological activity. Usually, this
relationship takes the form of an equation which also helps in eliminating the
luck factor from the drug design process. Classical QSAR approaches like
Free-Wilson and Hansch analysis have correlated the biological activity with
certain structural and physicochemical parameters, respectively.
Free and Wilson developed a mathematical equation to correlate certain
structural features (like the presence/absence of chemical substituents) with
bioactivity. It can predict the activity of only those compounds having known
substituents that have been included for developing the equation.
Hansch analysis
Hansch followed an extra-thermodynamic approach to develop a model in the form
of an equation. He proposed that biological activity can be correlated with
various physicochemical factors by a mathematical model.
Drug action involves two steps:
(i)
Transport of drug to the site of
action which mainly depends on lipophilic parameters like partition coefficient
and substituent hydrophobicity constant.
(ii)
Binding of a drug to the target
receptor which mainly depends on electronic (Hammett’s constant) and steric
parameters (Taft’s constant, Verloop’s steric parameter).
A typical Hansch equation looks like:
![]()
where biological activity is expressed as log 1/C
because of the very small value of “c”;
P
is partition coefficient;
σ
is Hammett’s constant;
Es
is Taft’s steric factor; and
k1,
k2,
k3,
k4 are constants. Hansch mathematical
equation helps in the prediction of new or unknown compounds; it also provides
information regarding the mechanism of the drug. However, the accuracy of this
model depends on various factors like accuracy of biological activity data,
inclusion of large dataset, and choice of appropriate parameters. A mixed
approach of both Free-Wilson and Hansch model has also been developed to widen
the applicability of both the methods. However, three-dimensional parameters
cannot be taken into consideration by any of the above two models.
Usually, the dataset is divided into training and test set molecules. A QSAR
model is built on a training set containing diverse chemical structures
including active and inactive molecules. The test set molecules are used for
testing the validity, predictive capacity, and accuracy of the developed QSAR
model. Currently, various multidimensional QSAR models like 3D, 4D, 5D, and 6D
QSAR have already been established based on multidimensional descriptors.
Molecular Descriptors Used in QSAR
Molecular descriptors are a numerical representation of chemical
information present within a molecule. There are many parameters such as
hydrophobic, electronic, and steric parameters, as well as associated
descriptors used for QSAR . Descriptors associated with hydrophobic parameters
are Partition coefficient (log P), Hansch’s substituent constant (π), hydrophobic
fragmental constant (f), distribution coefficient (log D),
apparent log P, capacity
factor in HPLC (log k, log kW),
and solubility parameter (log S). Hammett constant (σ,
σ+,
σ-),
Taft’s inductive (polar) constant (σ*),
ionization constant (pKa,
ΔpKa), and chemical shifts are
the descriptors used to define electronic parameters.
Similarly, steric parameters are defined by Taft’s steric parameter (Es), molar
volume (MV), Van der Waals radius and volume, molar refractivity (MR), and
Parachor. Atomic net charge (Qσ, Qπ),
super delocalizability, energy of highest occupied molecular orbital (EHOMO),
energy of lowest unoccupied molecular orbital(ELUMO) are known as quantum
chemical descriptors. Spatial descriptors such as Jurs descriptors, shadow
indices, radius of gyration, and principle moment of inertia are also used in
developing a QSAR model. The information about molecular descriptors depends on
the representation of a molecule and algorithm used for calculations of
descriptors.
3D-QSAR
The set of techniques that correlate biological activity with the three
dimensional structure of drugs is named three-dimensional QSAR (3D-QSAR).
The main objective of this approach is to identify spatial regions in the ligand
structure that are complementary to the receptor interaction site. A 3D-QSAR
investigation is based on the following assertions:
1.
It is the main compound that is being
modeled (not one of its metabolites) that is directly responsible for the
biological effect considered;
2.
The conformation being considered is
the pharmacophoric one;
3.
The interaction with the receptor and
the biological response that arises can be associated with only one
conformation;
4.
The site of action is the same for
the entire series of compounds examined;
5.
The biological activity is highly
dependent on enthalpic factors;
6.
Entropic contributions to the
interaction process are the same for all compounds;
7.
The system is supposed to be in
equilibrium, and kinetic factors are usually not taken into account, as well as
solvent effects.
The molecular alignment is crucial for the predictability of a 3D-QSAR model.
For flexible molecules, assertion 2 is not always true. Preferably, one should
take advantage of the maximum available information about pharmacophoric
conformation, rather than testing the various alignment methods that exist, such
as atom-to-atom alignment in a common substructure, alignment based on load and
mass distribution, or alignment based on genetic algorithm. The most used
3D-QSAR methods are CoMFA, HQSAR, and CoMSIA.
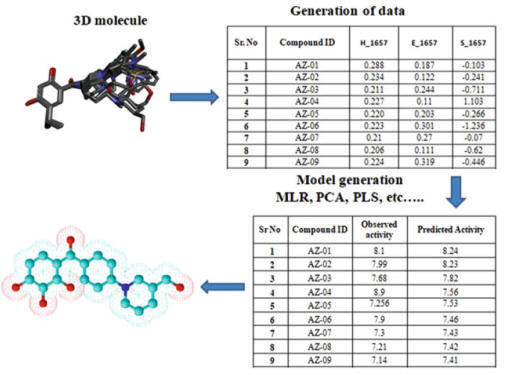
3D-QSAR generates the quantitative relationship between the
biological activity of a set of compounds and their 3D structural properties
(Fig.). 3D-QSAR uses a probe to determine values of 3D properties such as steric
and electrostatic of molecules and then correlate and build a relationship model
between 3D descriptors of molecules and its biological activity.
Molecular Shape Analysis (MSA)
MSA is an approach that includes conformational
flexibility and molecular shape data in 3D QSAR analysis. In MSA, the
3D structure of many compounds is superimposed to
find the commonly overlapping steric volume, and common potential
energy
fields between
superimposed molecules are also identified to establish a
correlation between the structure and activity of a set of compounds. This
analysis also provides structural insight into the shape and size of the
receptor-binding site.
Self-Organizing Molecular Field Analysis (SOMFA)
SOMFA divides the entire molecule set into actives (+) and inactive
(-), and a grid probe
maps the steric and electrostatic potentials onto the grid points. The
biological activity of molecules is correlated with steric and electrostatic
potentials using linear regression.
Comparative Molecular Field Analysis (CoMFA)
CoMFA is a grid-based 3DQSAR technique. It assumes that in most
cases, the drug–receptor
interactions are governed by non-covalent interaction. COMFA considers that a
correlation exists between steric and electrostatic
fields of molecules and their biological activity. Here, the steric
and electrostatic
fields of the ligands
at the various grid points in a 3D lattice are calculated. Partial least square
(PLS) analysis is used to correlate steric and electrostatic
fields with biological activities of molecules.
Comparative Molecular Similarity Indices Analysis (CoMSIA)
In COMSIA, molecular similarity indices serve as a set of
field descriptors. This technique of 3D QSAR is used to determine the
common features that are important for binding with the target molecule. Here,
not only steric and electrostatic features, but also hydrophobic
fields, hydrogen bond donors, and hydrogen bond acceptors are also
taken into account for predicting the biological activity of a compound.
3D
Pharmacophore Modeling
In pharmacophore modeling, the features governing the biological
activity are determined from a set of known drugs that binds to a specific target. The entire structure of a molecule is not responsible for
carrying out the biological activity. It is the only pharmacophore, which
decides the biological response. Pharmacophore modeling is used for searching
new potential drugs that share the same pharmacophore as available in other
biologically active drugs of the same target. Pharmacophore models are
hypothesis on the 3D arrangement of structural features such as hydrophobic
groups, aromatic rings, hydrogen bond donor, and acceptor. Structurally diverse
molecules bind with the receptor in a similar pattern, and their pharmacophore
interacts with the same atom or functional groups of the receptor molecule
(Fig.).
In the 3D QSAR model, molecules are aligned and superimposed with
the core structure, and the molecular descriptors are calculated based on their
conformation in the 3D space. The descriptors are correlated with biological
activity, and a mathematical model is established. The descriptors in the 3D
QSAR are the steric properties of the molecules, electrostatic forces, and force
field descriptors.

Advantages of 3D QSAR over 2D QSAR
1. No dependent on experimental values.
2. Can be applied to molecules of unusual substituents.
3. Not restricted to molecules of same structural class as in case
of pharmacophoric mapping.
4. Predictive ability.
QSAR APPLICATIONS
Quantitative-Structure Activity Relationship (QSAR) has been used
to predict therapeutic functions of molecules.
• QSAR improves compound libraries used in traditional HTS.
• QSAR can be used to direct combinatorial library synthesis
(libraries can be screened against biological targets of interest).
• QSAR has been applied to
de novo
drug design techniques when structural information regarding the
target is unknown.
The major dropouts of candidates during later stages of drug
development are due to their observed pharmacokinetic defects. Long-term and
tedious studies requires minimum of two years and costly. This cause delay and
cost accounting in the pharmaceutical industry. This demand for the prediction
tools.
QSAR has gained prominence in the prediction.
1. Quantitative-Structure Bioavailability Relationship (QSBR):
A subtype of
QSAR useful in analyzing the various parameters affecting the drug
bioavailability. A mathematical models correlates the molecular bioavailability
to structure descriptors.
2. Quantitative-Structure Metabolism Relationship (QSMR):
These models
are useful predicting the rate of cytochrome P450 mediated metabolism. This
model also predicts the rate of hydrolysis of prodrugs and soft drugs.
3. Quantitative-Similarity Toxicity Relationship (QSTR):
Toxic effects of clinical
candidates are the one of the major concern. This QSAR subtype is useful in
predicting the possible toxicity outcomes of the molecules through the
descriptor analysis.
Molecular docking
It starts with binding site prediction or identification. The binding site is
usually a concave region on the periphery of the protein where interaction with
the ligand occurs. Topological methods for identifying active sites, such as
CASTp,
allow us to study the suitable sites
for this interaction. Docking uses knowledge of protein binding sites to test
for interactions with small molecules.
Numerous positions are tested and then classified according to scoring criteria.
This scoring follows a mathematical function that varies depending on the
program and that distinguishes the different docking methods in their
applicability. The interaction energy is obtained through an empirical
expression that involves enthalpic, entropic, and hydrophobic contributions, in
addition to solvation effects. Docking can be rigid, which is useful when
scanning large databases of structures, or flexible, when we want to draw a more
detailed profile of the ligand-receptor interaction. The effects of the solvent
on the interaction can be simulated indirectly, through the introduction of a
correction in the expression of the interaction energy, or explicitly, through
the use of solvent boxes.
Molecular Docking Types
1. Rigid body docking:
In this docking procedure, the target protein and ligand
conformations are considered rigid. The conformational flexibility in the
molecular bond angle, bond length and torsion angle components are not
permitted. The substantial conformational change at molecular level is not
accounted in this type of docking, hence inadequate.
2. Flexible-ligand docking:
In this type of docking simulation, the target protein is
considered rigid (rigid conformation). The translational, rotational and
conformational degrees of freedom of the ligand can be explored.
3. Flexible docking:
This type of docking provides all degrees of freedom to the
target protein and ligands under investigation. It accounts complete
conformational change occurring at molecular level.
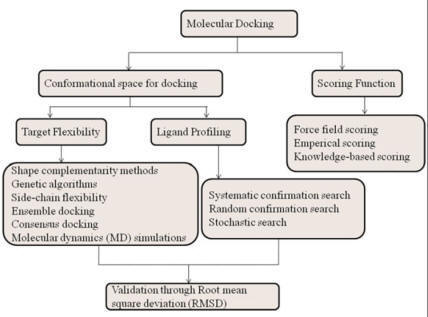
Different Types of Docking Based on Interactions
The selection of appropriate algorithms, tools, and parameters for
docking is an important challenge in molecular docking. In nature, different
types of molecular interactions such as protein–ligand (small molecule), protein–peptide like molecule, protein–protein, protein–nucleic acid, or
nucleic acids–ligand take place.
Different types of docking tools have been developed keeping in mind the nature
of interacting molecules, possible forces, and other parameters. In the
field of medicinal chemistry, ligand promiscuity is the topic of
discussion. Different folding patterns and structural arrangements were
deposited in large repositories like PDB, etc. The search for patterns and
similarities in binding sites and protein pockets allows the detection of
structural changes and behavior. Docking has been classified into many
categories based on the nature of the molecules involved in the interaction.
Protein–Ligand
Docking
Structure-based design is a very powerful approach to druggable
targets. Docking predicts the pose or orientation of a ligand on the binding
site of a target molecule or enzyme. For
flexible proteins,
protein-energy landscape exploration (PELE) is used for the correct assessment
of binding sites and poses. Through machine learning and molecular dynamics
using techniques, like self-organizing maps (SOMs) or
k-means
determines the
complementarity of protein and ligand conformations. For free energy
calculation, MTflex
uses Monte Carlo integration and
generating rotamers for binding residues based on low-energy values along the
free energy surface.
Protein–Peptide
like Ligand Docking
The peptide as a sample is highly variable due to high
flexibility. Nowadays, peptides are being used in the medicinal areas
proving their polypharmacological effects and suitability of protein–protein interaction.
It involves calculations that relate to confirmations and poses highlighted in Fig. Protein–protein interaction
networks can be perturbed by differential gene expression and disease mutations.
Molecular modeling approaches play an important role in optimizing the activity
of known peptide and also in designing the novel peptide as an inhibitor.
Protein–Protein
Docking
In protein–protein docking,
protein complexes are determined through sequence alignments, structural
comparisons, and multiple protein–protein interactions, within their defined confirmations and docking
positions. Protein structure initiative provides significant structural information for the community assessment of
structure prediction (CASP). For protein–protein docking and macromolecular interactions, critical
assessment of protein interactions (CAPRI;
http://capri.ebi.ac.uk) acts as a contest-space to challenge different human groups,
software, and servers into correctly predict the conformation of interacting
protein–protein pre-chosen
targets. Protein–protein docking can
be approached as a prediction for the whole complex minimizing each protein by
coarse grain models and using local search for the binding sites. Thus, the
major challenge
for protein–protein docking is
the
flexibility of the
backbone. For this reason, comprehensive computational studies need to be
conducted to successfully distinguish
realistic complexes from unrealistic predictions.
Protein–Nucleic
Acid Docking/Nucleic Acid–Ligand
Docking
Proteins and nucleic acids are the two main biological
macromolecules which act as a target for many processes/functions. Protein–RNA and protein–DNA interactions are
very important for replication, transcription, splicing, translation, and
nucleic acids degradation. Abnormalities in protein–nucleic acid
interactions are associated with a number of neurological diseases, cancer, and
many other metabolism associated issues. Protein–nucleic acid complexes are being solved by the researchers which
may help in understanding different interactions. NPDock is a protein–nucleic acid docking
tool and it uses the DARSRNP and QUASI-RNP statistical potentials for scoring
interactions of protein–RNA complexes. RNA
molecules have recently got an attraction as a drug target due to their
importance in biological key processes. However, as of now, the structure-based
docking that involves RNA molecules binding with a small molecule (ligands) is
not well established that lies under the protein ligand docking category.
LigandRNA is a scoring function used for predicting the RNA–small molecule interactions. It is based on a grid-based algorithm,
and a knowledge-based potential for scoring is derived from sites of
ligand-binding interactions in the known RNA–ligand complexes. LigandRNA takes RNA receptor
file and ligand pose
file as an input and
provides the ranking poses consistent with their score as an output. The modified version of Dock6 also includes RNA–ligand docking
facility. Ligand–RNA docking related
problems were solved by incorporating classical molecular mechanics force
field for calculating the interaction between the RNAand ligand.
Molecular Docking Steps
A
typical docking procedure consists of four steps: ligand setup/preparation,
protein setup/preparation, docking, and postdocking analysis.
Ligand preparation
The input file format of a chemical structure is very important because it
represents the atomic coordinates, bond types, and bond order of a ligand.
Docking results along with the molecular interactions may get affected by any of
the parameters of the ligand, i.e., protonation state, tautomer, conformer, etc.
Therefore, ligand structure is converted into 3D conformation and then refined
by energy minimization using molecular mechanics protocol.
Protein preparation
A high-resolution X-ray crystallographic structure of a protein is usually
preferred over other structures. In case of nonavailability of X-ray structure,
the protein structure can be generated from homology modelling also. When
several X-ray structures of the protein are available, holoenzyme (with ligand)
complex structure is preferred over apoenzyme (without ligand). If many
holostructures are available, the one with a cocrystallized ligand at the
binding site is preferred. Hydrogens are added to the protein structure,
especially
polar hydrogens for optimizing the hydrogen bonding network. Missing side chains
or residues should be checked and corrected before the final refinement of
protein, in which it undergoes minimization to remove any clashes. The binding
site information is obtained from the cocrystallized ligand complex at the
binding site of the enzyme. It helps in confining the 3D space of the binding
site into a grid box, where a suitable binding pose of the ligand is searched.
In the case of apoenzyme structures or when the binding site information is
unknown, a time-consuming blind docking is performed where the entire protein
surface is scanned for a suitable binding site. Currently, many programs are
available which can detect the binding sites within an apoenzyme structure based
on various pharmacophoric features.
Docking
The pioneering work of Kuntz et al. has led to the development of many
open-source as well as commercial docking programs. It is comprised of a search
algorithm and a scoring function that critically determines the speed and
accuracy of docking. It searches and ranks various poses of ligands inside the
conformational space available within the binding site of the receptor.
Validation of the docking program is carried out by satisfying various
benchmarking parameters to prove its speed, accuracy of prediction, and ability
to distinguish actives from inactives.
Binding free energy calculations
Binding free energy is an important aspect of ligand to find fitting within a
binding site and achieving the global energy minimum for the complex. Thus, free
energy is of crucial importance as they regulate
binding affinities, protein folding. And as fundamental thermodynamic quality in
binding, folding, and reaction kinetics, it is important to quantify binding
free energy in computational techniques.
The binding free energy accompanying the binding of a ligand to target protein
is determined by the following equation:

Postdocking analysis
The top poses are ranked by the least binding energy score (usually with a
negative sign). Poses which show steric or electrostatic clashes may be screened
by applying topological filters.
Energy minimization of a ligand pose inside the binding site can be
performed by another program
and analyzed by machine learning methods.
Scaffold enrichment may be applied on
the hit molecules as an alternative refinement method to recover false
negatives, if they share a common structural framework with true positive
ligands.
Based on the flexibility of ligands and receptors, docking can be of two types:
rigid docking and flexible docking. When both the receptor and ligand are kept
rigid, a limited search space is available encompassing only three
rotational and three translational degrees of freedom. A predefined set of
ligand conformations can be used to address the ligand flexibility. Various
scoring functions like Monte Carlo simulations, simulated annealing,
evolutionary, and genetic algorithm methods have been used to incorporate ligand
flexibility, whereas the receptor is kept rigid. Molecular dynamics (MD)
simulations are used to treat receptor flexibility, but they consume much
computational resources and time.
Analysis of
Docking Results
Analysis of docked complex structure obtained from molecular
docking study is one of the essential tasks for visualization of protein–ligand interaction
at the atomic level using molecular modeling tools in 2D or 3D. In this analysis
we can identify the number of hydrogen bonds formed among a different functional
group of the ligand with amino acid residues present in the binding site of
protein along with their bond length, because hydrogen bonding plays a significant role in protein–ligand interaction.
Besides, we can also analyze hydrophobic and cation–pi interaction. This
analysis facilitates researcher to choose the best interacting ligand because in
some cases, the binding energy of two or more ligands is the same but the number
of interacting amino acid residues are less or more. Therefore, in such a
situation, generally we choose ligand having more interaction
with the target in terms of interacting amino acid residue numbers.
PyMOL and Chimera are widely accepted tool analyses of docking results by
selecting different poses of ligand generated during molecular docking and
visualization of interacting residues in 3D. Besides, LigPlot is one of the
highly cited and recommended tools for analysis of docking results in 2D format.
Validation and Accuracy
Docking accuracy is usually assessed by the ability to reproduce
the experimentally determined binding mode of a ligand. The predicted binding
mode is chosen based on the best-scoring protein-ligand complex. Docking time
will increase linearly with the number of structures used. The success rate in
retrieving binding modes of known protein-ligand complexes is an important
validation for docking programs. The success of the docking is measured by root
mean square deviation (RMSD) between the experimentally observed heavy-atom
positions of the ligand and those predicted by the program. Poses with an RMSD <
2 Å are considered a success, and docking whose RMSDs lie between 2 and 3 Å are
considered as partial success. The evaluation of the docking results relies
mainly on a scoring function that ranks the bonding poses according to specific
properties. State-of-the-art docking programs correctly dock ~70-80% of ligands
when tested on large sets of protein-ligand complexes.
Absorption, distribution, metabolism, excretion, and toxicity
(ADMET) prediction
The determination of pharmacodynamic, pharmacokinetic, and toxicological
properties is essential for obtaining relevant bioactive compounds. Lipinski’s
rules
(a molecule with a molecular mass
less than 500Da, no more than 5 hydrogen bond donors, no more than 10 hydrogen
bond acceptors, and an octanol-water partition coefficient log P not greater
than 5) allow determining whether the compound has potential as a drug without
presenting toxicological characteristics that make its use unfeasible. Many
tools for ADMET are available, such as QikProp.
ADME analysis and measures of drug-likeness
Usually, 9 out of 10 research projects in drug discovery course face end-stage
failure. These 10 projects involve the synthesis of about 10,000–20,000
molecules, followed by their activity studies. The failure rate of the drug
discovery process in the pharmaceutical industry is too high, usually about
99.99%.
These failures come with a huge price
of approximately
$500
million and
$2billion.
Many candidates undergo failure in
late-stage clinical studies due to poor ADME (absorption, distribution,
metabolism, elimination) properties.
Therefore, various computational models were developed to predict ADME
properties before clinical studies. It helps in the preselection of good
drug-like candidates for synthesis and activity studies, reduces the failure
rate and cost involved in a clinical trial by removing compounds with bad ADME
profile, and improves the understanding to correlate experimental and predicted
ADME parameters.
Gradually, various drug transport
models like Caco-2 cell permeability representing intestinal absorption were
also incorporated. Eventually, the toxicity profile gets coupled with other
ADME-related properties to develop ADMET parameters which have been successfully
used in virtual screening procedures for filtering large databases to select hit
molecules. Data obtained
from high-throughput in vitro screening assays are used by numerous
computational methods and descriptors to build ADMET models.
Various molecular property-based descriptors like polar surface
area, hydrogen bondingnetwork, octanol/water partition coefficient, or
semiempirical-based descriptors can help in the quantification of
pharmacokinetic or ADME properties, which is further correlated with its 3D
structure by suitable models.
Measures of drug-likeness
Qualitative evaluation of basic descriptors is the most common way for examining
the ADME profile of a molecule, as proposed by Lipinski’s “rule of five.” A
compound is considered to have poor absorption if it violates any two of the
proposed parameters (molecular weight not more than 500; number of H-bond
acceptors and donors not more than 10 and 5, respectively; calculated logP
not more than 5).
Researchers subclassified the
descriptors based on oral and nonoral drugs, different target diseases to
examine their effects on the ADME parameters. Vieth et al. evaluated 1729
marketed drugs and reported a statistically significant difference between
injectable drugs (high molecular weight, more polar) and oral drugs (low
molecular weight, less polar). Moreover, the pharmacokinetic parameters for
injectable drugs were more flexible as compared to oral drugs.
It led to the conclusion that pharmacokinetic parameters need
biased property distributionas per different targets and routes of
administration.
Aqueous solubility and lipophilicity
Aqueous solubility is an essential parameter to be predicted for molecules
targeting the oral route of delivery. Good solubility is highly necessary for in
vitro,
in vivo assays and for predicting the
absorption in the gastrointestinal tract. Poor solubility negatively affects
absorption and assay results and increases the development cost. Therefore,
various quantitative structure-property relationship (QSPR) models have been
established for the prediction of aqueous solubility using numerous molecular
descriptors. Since the composition of gastrointestinal fluids is not taken into
account, aqueous solubility cannot be considered as an optimal model for
predicting solubility.
Yalkowsky and Jain have developed an
in silico model called “general solubility equation” to predict aqueous
solubility with good accuracy.
Lipophilicity of a drug helps itself in getting dissolved in lipid
phase, and thus it can pass through the bilayer lipid membranes in the
gastrointestinal tract, which can be predicted by the descriptors: LogP
(octanol/water partition
coefficient), LogD
(distribution coefficient), and chromatographic hydrophobicity index.
Ionization state
Represented by pKa,
it affects the solubility, lipophilicity, and permeability of a compound and
thus becomes essential for good absorption of oral drug candidates. Hammett and
Taft’s approach, semiempirical methods, and density functional theory are some
of the methods which are used for the prediction of ionization state.
Permeability
Transport of a drug across the membrane by a passive method is referred to as
permeability. Apart from the in vitro determination of partition coefficient and
distribution coefficient, several cell line-based assays have been developed.
Caco-2 (human colorectal carcinoma) cell line can be modeled to predict
intestinal permeability of drugs. This model has been utilized in the early
stages of drug discovery for ranking compounds based on absorption and
permeability. Similarly, MDCK (Madin-Darby canine kidney) cell-based assay can
also be modeled to predict permeability as well as drug-receptor interactions.
Blood-brain barrier
Both the aqueous solubility and lipophilicity of a compound determine its
capability to penetrate the blood-brain barrier by passive diffusion. It was
measured by the parameter LogBB, determined by the ratio of concentration of
drug in brain to concentration of drug in blood. Currently, several models have
been developed for the accurate prediction of LogBB by using various machine
learning methods.
Distribution
Volume of distribution can be predicted by various in silico models, which
correlates the lipophilicity and solubility descriptors with free and bound
fraction of drug with plasma proteins. Amo et al. have established a model to
estimate the volume of distribution whose accuracy was comparable with
commercial counterpart Volsurf+.
Metabolism
Various in silico models can predict the site of metabolism along with its
substrate nature against a specific metabolic enzyme. A vast dataset of diverse
chemicals can be taken to generate models and then converted into online
prediction tools for determining pharmacokinetic parameters related to
metabolism, e.g., fast metabolizer, SMARTCyp.
Excretion
Kusama et al. have developed a chemoinformatic-based model based on molecular
weight, lipophilicity, charge, and protein-bound fraction in plasma. It helped
in the prediction of major clearance pathways of 141 drugs with good accuracy.
However, lack of a larger experimental dataset is a major obstacle in the
development of more accurate ADME models. Yet, computational chemists strive to
develop good predictive ADME models to assist the drug discovery project.
ADMET screening
ADMET is the short form for absorption, distribution, metabolism, elimination,
and toxicity. In the course of drug designing processes for the period of last
two and half decades, in silico ADMET analysis has become a very useful means of
cost-effective tool as the ADMET properties are accountable for the
nonfulfillment of 50% or more of drugs in the clinical stages. ADMET analysis of
drugs is principally focused on computational pharmacokinetics and toxicity
modeling. Conventionally, in the drug development pipeline, the ADMET tools are
applied at the end stages, but in the recent time, it is applied at an early
stage of the drug development procedure owing to the easy accessibility of in
silico ADMET tools which readily discard molecules with poor ADMET at the early
stages of the development, leading to substantially cost-effective drug
development process.
Some of the commonly available tools
which are involved in the monitoring of ADMET characteristics are ADMETlab,
admetSAR, SwissADME, PreADMET, eADMET, and Tripod.
ADMETlab
It is a Web-based tool that broadly relies on the incorporation of a database
collected from the existing ADMET and several basic endpoints associated with
possible physicochemical profiles to facilitate the ADMET evaluation. There are
four main considerable components that allow users to properly evaluate ADMET
properties, which are:
(1)
Assessment of drug-likeness profile
using rules of five and one prediction model.
(2)
Prediction of ADMET properties using
31 endpoints including,
·
3 on basic property
·
6 on absorption
·
3 on distribution
·
10 on metabolism
·
2 on elimination
·
7 on toxicity
(3)
Evaluation of efficient ADMET for
single chemical entity and.
(4)
Similarity searching and comparing
against 288,967 entries in the ADMET database.
The design of ADMETlab is created based on Python’s Django framework and is
freely available online at
http://admet.scbdd.com/.
admetSAR
It is an open-access tool containing a database associated with ADMET properties
that constantly update by collection, curating, and management of available data
related to ADMET properties from the existing various published literature
studies. In admetSAR, for an exclusive collection of over 96,000 compounds there
are ADMET annotated data points of 210,000 or more, with 45 different types of
ADMET-related properties. The admetSAR database can be easily accessed by a
user-friendly interface using CAS registry number, common name, or structure
similarity to enquire a definite chemical’s ADMET profile. To predict ADMET
characteristics of novel chemicals with high accuracy, the database includes
·
22 qualitative classification and
·
5 quantitative regression models
To safeguard its utility and quality, the database is updated every month or
quarter with the addition of high-quality published data associated with
toxicity. AdmetSAR is available at free of cost from the Web site:
http://www.admetexp.org.
SwissADME
It is a freely available Web tool that allows the prediction of pharmacokinetic
profile, drug-likeness, and medicinal chemistry suitability of a compound; it
also contains in-house proficient methods such as the BOILEDEgg, iLOGP, and
Bioavailability Radar.
It is easily accessible from the login-free Web site
http://www.swissadme.ch.
Compared to the other freely available Web-based tools for ADME and
pharmacokinetics screening such as pk-CSM and admetSAR, the strong points of
this tool are:
·
Comprising different input methods.
·
Ability to compute several molecules.
·
For each individual molecule option to display, save, and share
outcomes.
Toxicity prediction
During drug discovery and development, identifying molecules with the best
possibility to become a clinically useful compound, candidates are evaluated
according to diverse parameters to direct the selection and promotion of
chemicals for synthesis and test. It is important that the molecules must
display high biological activity along with low toxicity. Since more than 50% of
the candidates failed in drug development stages owing to toxicity, a set of
toxicity screening has been implemented in most pharmaceutical establishments to
counter this failure at the development with an aim of discarding compounds that
are likely to fail further down the line in the discovery phase.
Some of the available tools for toxicity screening of
compounds are pk-CSM and
PreADMET.
pk-CSM
pk-CSM is a Web-based, freely available tool for the analysis and optimization
of pharmacokinetic and toxicity properties. It assists medicinal chemists to
discover the balance between safety, effectiveness, and pharmacokinetic
properties. The 30 inbuilt predictors are divided into five major classes:
·
predictors based on absorption, which contain seven predictors
·
predictors based on distribution, which comprise four predictors
·
predictors based on metabolism, which comprise seven predictors
·
predictors based on excretion, which comprise two predictors, and
·
predictors based on toxicity, which comprise 10 predictors
The tool predicts small molecules’ pharmacokinetic and toxicity profiles. The 10
predictors in pk-CSM based on toxicity, which predicts toxicity profiles of
compounds, are:
·
Maximum recommended tolerated dose (MRTD)
·
Oral rat acute toxicity (LD50)
·
Oral rat chronic toxicity—lowest observed adverse effect (LOAEL)
·
T. pyriformis
toxicity
·
Flathead minnow toxicity (LC50)
·
hERG I inhibitor
·
hERG II inhibitor
·
AMES toxicity
·
Hepatotoxicity
·
Skin sensitization.
pk-CSM is freely available online from the Web site:
http://structure.bioc.cam.ac.uk/pkcsm.
PreADMET
PreADMET
is a Web-based tool for the analysis
of ADME and toxicity data. The functions of the tool can be divided into four
parts:
(i)
Molecular descriptors calculation;
(ii)
Drug-likeness prediction considering
well-known rules;
(iii)
ADME prediction; and
(iv)
Toxicity prediction.
In this tool, toxicity prediction of a compound is carried out by two
predictors: Ames test and rodent carcinogenicity.
Ames test
It is conducted to assess the mutagenicity of a compound. It predicts molecules’
toxicity profile by comparing the data of NTP (National Toxicology Program) and
US FDA, which are the outcomes of the in vivo carcinogenicity tests of mice and
rats for 2years.
Molecular dynamics
Molecular dynamics can be defined as the set of experimental protocols that
simulate the conformational variations experienced by the molecule by the action
of forces acting on the medium. This procedure applies Newton’s equations to the
internal coordinates of the molecule, within a given time frame, in order to
follow the variations in the internal degrees of freedom of the molecule. The
differential form of Newton’s classical equation Fi =
mi ai. An error inherent to the method will be more pronounced the longer the
time step used, which is typically on the order of 0.5 to 1 femtosecond (1 fs
=
10-15
s). In general, we start with a minimized structure and process the dynamics,
recording the resulting structure from time to time and minimizing each of these
conformations again, so that, in the end, we proceed to select the conformation
whose energy no longer varies.
Molecular dynamics (MD) Process
During molecular interactions, the receptor and especially its binding site can
undergo conformational changes, which affects the binding energy as well as
stabilization of the ligand-receptor complex. Receptor flexibility has been
overlooked by docking methods to achieve speed but compromising with accuracy.
Molecular dynamics has been successfully employed for simulation of
ligandreceptor binding, conformational sampling, and accurate prediction of the
energetics of the system. For simulating the movement of each and every atom of
the ligand and receptor, we need to quantify the velocity as well as the force
acting on the atoms. Initial potential energy gives information about the
coordinates, energy, and velocity for each atom of the system. Applying a force
on each atom for a very short span of time (approximately in femtoseconds), we
can determine the acceleration from the Newtonian equation of motion, and
subsequently velocity and coordinates at a new position for each atom.
This process is repeated to get new positions of atoms
with respect to the applied force and gradually takes the shape of a trajectory.
Once the trajectory is defined, we can simulate the motion of an atom a short
time into the future. The new position of the atom at a specific time in the
future can be determined from its initial position coordinates and velocity by
solving the equation known as Taylor’s expansion. Then, energy minimization of
these structures is carried out by molecular mechanics which also helps in the
study of conformation and energetics.
Two important factors like the temperature of simulation and time steps are
critical for carrying out MD simulations. Application of high temperature during
simulation helps in overcoming the energy barriers, so that the conformations do
not get trapped in local minima and can reach global minima. The selection of a
small yet proper time step is essential for searching all the possible
conformations. In short, molecular dynamics is referred to as solving the
Newtonian equation of motion for all the atoms of the system as a function of
time.
A common protocol for MD simulation involves the following steps:
Target structure:
A 3D receptor structure (good resolution), determined by the NMR method or X-ray
crystallography, is preferred for only receptor simulation. It can be downloaded
from the protein data bank (http://www.pdb.org). In the case of ligand-receptor complex simulation, usually a
docking output file is used as input for MD simulations where the ligand is
present at the binding site.
Input structure:
Topology parameters are generated for all the atoms present in the system which
contains the necessary information about the atoms, bond connectivity, angles,
their coordinates and velocities, etc. Required hydrogen atoms are added, and
protonation states, terminal residues, and disulfide bridges are checked.
Setting the simulation environment: A simulation box (similar to the grid box in docking) is created
around the binding site and is immersed into a periodic box of water molecules
to solvate the protein. The water molecules are represented by various in silico
models like simple point charge (SPC) or extended simple point charge (SPC/E),
or three-point (TIP3P), or four-point (TIP4P). A proper dielectric constant and
force field is selected along with the addition of required counterions to set
the simulation environment.
Energy minimization:
Before MD simulation, a short energy minimization (usually 500 iterations with
the steepest descent algorithm) is carried out. It helps in relaxing the
structure and also removes high-energy artifacts like broken hydrogen bonds
which can distort the entire system.
Heating up the system:
A high temperature of 1000K is applied for 1–25 picoseconds to stabilize or
equilibrate the core structure.
MD simulation:
The simulation period is set up as per the protein size and the availability of
computational resources. The time interval is also set up, at which the output
coordinates of the system are recorded for further analysis. For example, a
simulation is run for a period of 200 picoseconds with a sampling time of 1
picosecond. Different conformations were recorded at 1 picosecond interval and
confined the total number of structures to 200 frames.
Trajectory analysis:
All the conformers were retrieved, and energy was minimized to rank the
structures with the lowest energy binding modes. The stability, as well as the
structural integrity of the system, is determined by measuring the
root-mean-square deviation of all heavy atoms with respect to the parent
structure. Free energy of binding can also be calculated to compare the
stability of the complex before and after the simulation.
Molecular dynamics simulations are helpful in the identification of cryptic
binding sites, allosteric binding sites, binding pose of ligand, and accurate
estimation of binding affinity. MD simulations have also been used in virtual
screening for allowing receptor flexibility during screening. This method is
known as a relaxed complex scheme (RCS) where MD simulation is run on the
receptor structure to obtain multiple conformations, with which the potential
hit candidates can be docked. Therefore, every hit candidate is associated with
a series of docking scores and can be ranked based on the average docking score
over a receptor. The development of new force fields, conjugation of quantum
mechanics, and upgraded computational resources have significantly improved the
performance and applications of MD simulation.
Current Tools for MD Simulation
Several tools are available to investigate the atomic-level changes
in the biomolecules using the MD simulation method. Some provide the graphical
user interface like Desmond, while some run in command lines like GROMACS and
AMBER. Some famous and widely used tools for MD simulation are GROMACS, AMBER,
Nanoscale MD (NAMD),
and CHARMM-GUI. For running such MD simulations, increased hardware
power and software are essential components.
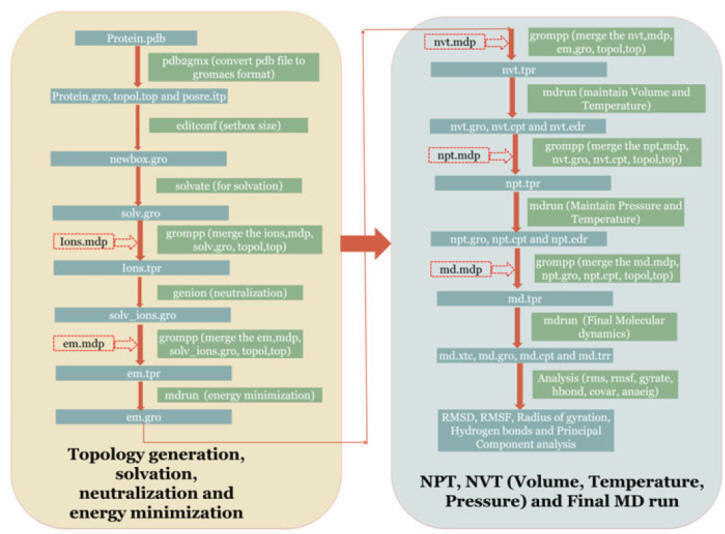
Recent Advances in Hardware to Run MD Simulation
Rapid development in computer hardware is a crucial part of MD
simulation. Two reasons have an impact on trajectory analysis. The
first one is the run of the long simulation result in GBs to TBs data
storage, and the other is to develop the new rendering engines for the
visualization effects using the latest video chipsets. Due to the advancement in
the computer hardware, simulations can be performed from ns to
μs with the help of GPUs (graphics processing units) that is configured with the molecular simulation suite. The GPU cards are
replacing the CPU (central processing unit) and becoming commodity software and
play a crucial role in decreasing the time for MD simulation. The CUDA (Compute
Unified Device
Architecture) is a newly invented parallel computing platform, and its use in
GPU increases the number of cores to run a long simulation within time. Due to
the emergence of GPU-CUDA technology, vigorous and massively parallel clusters
are developed, such as special purpose supercomputer Anton and Blue waters. They
are precise for running the MD simulation of biomolecules from
μs to ms time-scale.
But such
resources are limited for limited researchers. To remove the
time-scale gap, there is an urgent need to develop newer algorithms that allow
enhanced sampling in the defined areas of
conformational space and access long time-scale actions using necessary
hardware. The purpose of this algorithm is to collect sufficient sampling that could result in the Boltzmann distribution of
the diverse conformational states for the accurate calculation of the
thermodynamic and kinetic properties of the system. By the modification of the Hamiltonian method is to add a bias potential,
several approaches have been developed like hyper dynamics, local elevation, and
accelerated MD. In the case of hyper dynamics simulation, the identification of transition state required, but it is not necessary for
classical MD simulation. Several tools are available to perform the MD
simulation study with CPU or GPU. A few of the widely used tools are described
in brief below.
GROMACS
GROMACS is the most widely used software for MD simulation. It is a
freely available tool, and a brief tutorial of this tool can be accessed by this
link (http://www.mdtutorials.com/gmx/) (Pronk et al.
2013).
In the GROMACS
simulation kit, MD simulation can be performed at various temperatures and pH
values. In this simulation tool, several commands are available to perform a
distinct function and calculate specific structural
parameters. GROMACS, which is one of the MD simulation software, can read only
the 20 natural amino acids, i.e., the non-standard amino acids are not read by
GROMACS algorithms. Sometimes, there are force
field limitations, for
instance, Gromos and Amber cannot read the nicked DNA, but the same force
field can read the same non-nicked DNA. The brief methodology for MD
simulation using GROMACS is shown in Fig.
To start, the user creates a box and
fills the solvent (water). The solvent model depends on the force
field. After placing the protein in the defined box in the solvent, the charge of the system is neutralized
either by the addition of Na+ or Cl_ions; this is
followed by the minimization of the system using the steepest descent method.
Then, NVT (the constant Number of particles, Volume, and Temperature) simulation
is run to maintain the volume and temperature of the defined system. The temperature of the system arises from 0 and attains
the desired temperature that is set by the user. After that, NPT (the constant
Number of particles, Pressure, and Temperature) simulation is run to maintain
the pressure of the defined systems.
Several parameters are set by the addition of the .mdp
file. Finally, MD simulation is performed that provides the
coordinates of each step in the form of a trajectory. The trajectory can be
analyzed by using various tools that are embedded in GROMACS, like gmx–rms, gmx–rmsf, gmx–gyration, and gmx–hbond. These data
can be plotted in an interactive form by using GRACE (Graphing Advanced
Computation and Exploration of data), a Linux based software. For example, a
water embedded protein molecule, placed in a box visualized by VMD (Visual
Molecular Dynamics) shown in Fig.
AMBER
AMBER simulation suite is a collection of programs that are used to
carry out and analyze the MD simulations for proteins, carbohydrates, and
nucleic acids. Three main components of the AMBER tool are preparation,
simulation, and analysis. The Antechamber and LEaP are the main program for the
preparation of macromolecules. The Antechamber tool prepares the
files into the force
filed descriptor
files, which is read by the LEaP program for molecular modeling. The
LEaP program then creates the topology
files and Amber
coordinates, which is then used in the MD simulation. The Sander program
performs the MD simulation by
fixing the
temperature, pressure, and pH of the defined system. Lastly,
the analysis part is performed by the ptraj module, which calculates the RMSD,
RMSF, radius of gyration (Rg), H-bonds, and cross-correlation functions.
CHARMM-GUI
CHARMM-GUI is a simulation tool for the analysis of macromolecular
dynamics and associated mechanical attributes. It performs standard MD
simulations by using state-of-the-art algorithms for time stepping, long-range
force calculation, and periodic images. Various analyses, such as energy
minimization, crystal optimization, and normal mode analysis, can be performed
using CHARMM.
NAMD
The simulations of much large biomolecular systems are performed
using NAMD. The NAMD is available free of charge. The source code documentation
including a set of compiled binary
files configured with various parallel source software for calculations are
freely available to the user. It supports massively parallel CUDA technology.
NAMD can be used with graphical user interface software VMD. The simulation can
be set and analyzed using the VMD as an interface. It is also compatible with
AMBER and CHARMM.
Drug-likeness
Drug-likeness is a qualitative concept used in drug design. The
molecules with inherent physicochemical and therapeutic features are termed as
drug-like. The phenomenon is known as drug-likeness. Drug-likeness analysis
generates predictive models for optimizing the pharmacokinetic properties. This
analysis filters the compound libraries to remove unlikely molecules from the
consideration. Neural network models categorized 83% of Comprehensive Medicinal
Chemistry (CMC) database molecules and ~65% of MDL Drug Data Report (MDDR)
molecules as drug-like. It also clustered 73% of compounds from Available
Chemical Directory (ACD) as non drug-like compounds.
Drug-Likeness
Analysis Tools Online (Freeware) Tools
Molinspiration:
An online tool for the molecular
property calculation and biological activity predictions. The molecular
properties namely molecular weight, log P, polar surface area (PSA), number of
rotatable bonds, molar volume, number of hydrogen bond donors and acceptors
(HBDs and HBAs) can be predicted. It also predicts the molecular affinity for
the targets namely GPCR, ion channels, nuclear and kinase receptors. Molecular
structure can be manipulated and processed till arriving with optimal
characteristics.
Website:
http://www.molinspiration.com/
Molsoft:
It provides molecular property
prediction, structure prediction, binding site prediction, drug target ranking,
2D to 3D conversion of molecules, data set clustering, QSAR model building, 3D
pharmacophore construction and search, and molecular visualization services.
Website:
http://molsoft.com/mprop
Chemicalize.org:
A free web-based drug-likeness
tool from ChemAxon. It has tools namely calculation, structure search, document
search and web viewer. The query structures can be submitted using MarvinSketch
Java applet. The molecular properties such as polarizability, log P, log D and
pKa can be calculated.
Website:
http://chemicalize.org
PASS;
Prediction of activity spectra for substance:
It is a
software application (available online and download) to predict the biological
activity spectra of drug-like molecules. It estimates the biological activity
profile of virtual molecules based on their structure. It also predicts
carcinogenicity, mutagenicity, teratogenecity and embrotoxicity informations.
Website:http://www.way2drug.com/PassOnline/predict.php
PreADMET:
A web-tool for drug-likeness
analysis and absorption, distribution, metabolism and excretion (ADME)
predictions. It also supports in toxicity predictions and molecular
visualization. A commercial tool (PreADMET 2.0) also available.
Website:
http://preadmet-bmdrc.kr/druglikeness
ALOGPS2.1: Virtual Computational Chemistry
Laboratory (VCCLAB) features the ALOGPS2.1 (interactive on-line predictor) for
predicting molecular water solubility, log P, pKa, Log D, Log W and Log S
values. It is developed based on the associative neural network (ASNN).
Website:
www.vcclab.org/lab/alogps
Lipinski’s rule
The impression of physicochemical assets confines the solubility and
permeability of drugs introduced during Lipinski’s analysis of the Derwent World
Drug Index. Lipinski et al. confirmed that orally administered drugs are
expected to feature in parts of chemical space distinct by a restricted range of
molecular characteristics. The criteria of physicochemical properties for
Lipinski’s rule are mentioned in
Table.
Physicochemical properties for Lipinski’s rule.

The term Lipinski’s “rule of five” (RO5) originated from the molecular
properties of drugs which are found to be multiples of five. In this method,
drugs and nondrugs molecules are distinguished by a definite range of
physicochemical accounts and molecular characteristics.
Veber rule
Veber et al., in a paper released in 2002, proposed the following:
Irrespective of molecular weight, the important predictors for good oral
bioavailabilityare lowered molecular flexibility, which is estimated by the
extent of rotatable bonds, and low polar surface area or total hydrogen bond
tally including both donors and acceptors. In the extensive set of data,
filtering compounds with poor oral bioavailability from those with satisfactory
value, the frequently functional molecular weight
endpoint at 500 does not itself suggestively separate compounds.
According to the rule, the criteria for
compounds with good bioavailability are mentioned in
Table.
Veber rule’s criteria for compounds.

Teague Rule of Three (RO3):
Teague et al described the
required molecular features of drugs. According to the rule (RO3), the molecules
with the molecular weight in the range of 100 to 350 Da and clog P in the range
of 1 to 3 are predicted to have better physicochemical properties.
Oprea
Rule of Three:
Oprea conducted the study on MDDR,
CMC, Current Patents, Fast-alert, New Chemical Entities and ACD to assess the
parameters responsible for bioavailability. According to the study the following
molecular characteristics are essential for promoting the drug bioavailability.
• Number of
rings, > 3
• Number of
rigid bonds, > 18
• Number of
rotatable bonds, > 6
Norinder Rule of Two (RO2):
Norinder rule describes the
required molecular features for crossing the blood-brain barrier (BBB). This
rule accounts the nitrogen and oxygen atom count.
• Rule 1: The sum of nitrogen and oxygen atoms (N+O) should be
less than (<5). It indicates the better molecular BBB permeation
characteristics.
• Rule 2: The calculated log P value should be more than 0 (> 0).
The number obtained by subtracting the sum of nitrogen and oxygen
atoms from log P is also indicative of better BBB permeability. These rules
cannot explain the pharmacodynamic nature of the molecules. Peptidomimetics,
transporter substrates and natural products do not obey these rules, possibly
due to transporter effects. Drug-likeness filters recognize the compounds which
resembles with existing drugs. But the new class compounds can not be identified
and is a major challenge.
Traffic lights:
Lobel et al., modified the RO5,
based on the rules generated by other teams (Veber, Ghose, Wenlock, Monika).
These molecular parameter prioritizations are known as traffic lights (TLs).
• Molecular weight, < 400 Da
• Log P, < 3
• Solubility at pH 6.5, < 50 mg/L
• Polar surface area (PSA), < 120 Å2
• Rotatable bonds, < 7
PhysicoChem scores are calculated to know the
in
silico
prediction correlations. The score
ranges from 0 to 1. The lower score is indicative of good
in
silico
correlation.
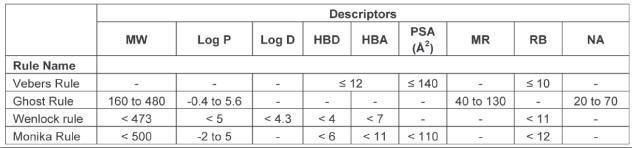
Conclusion
CADD has immensely helped medicinal chemistry researchers to bypass or hasten
multiple procedures in the drug design and discovery to find out potent clinical
candidates in a short period of time. CADD is very useful in critical steps such
as the hit-to-lead discovery and lead optimization; therefore, it paves the way
for time as well as cost deduction. Discovery through creating three dimensional
structures of ligand and protein, simulation, prediction of binding interactions
and energy would be a very tedious and time-consuming task. However, as compared
to conventional drug design and discovery, CADD has several advantages and is
classified into SBDD and LBDD. Docking, molecular dynamics, and pharmacophore
modeling are the essential steps in SBDD. Similarity search, QSAR model, and
pharmacophore modeling are part of LBDD. In
the drug discovery paradigm, establishing the drug-likeness with the assistance
of Lipinski’s rule of five or Veber’s parameters or rule of three could be a key
approach, which determines the drug-like candidates in a reasonable quick
timeline. The different in silico models predict various parameters of lead
compounds, aqueous solubility, lipophilicity, ionization state, permeability,
distribution, metabolism, and excretion. Hence, a detailed pharmacokinetic
profile obtained from in silico methods would facilitate a robust approach in
drug design, discovery, and development. Nonetheless, we have many innovative
techniques in medicinal chemistry, and the discovery of advances must be
encouraged to further reduce cost and time duration.
Fragment-based drug design
Fragment-based drug design (FBDD) is a biochemical and biophysical method to
detect very small molecules or fragments, which can bind to specific targets and
help in engrossing drug molecular leads.
Generally, it starts with screening and identification of low
molecular weight compounds with fewer convolutions for binding to a specified
target.
The fragments must be small in size to avoid unsuitable interactions, i.e., the
identified fragments generally show identical binding affinity, which is
advantageous for further optimization.
Also, as the fragments are very small for binding to targets and
thus after identification to enhance the binding potential of fragments, further
strategies like fragment linking, merging, and growing can be applied.