AUTOIMMUNITY
The state of adaptive immune system responsiveness to self antigens that occurs when mechanisms of self-tolerance fail was defined as autoimmunity. A disease caused by a breakdown of self-tolerance such that the adaptive immune system responds to self antigens and mediates cell and tissue damage known as autoimmune disease. Autoimmune diseases can be organ specific e.g., thyroididits or systemic e.g., systemic lupus erythromatosus. An antibody produced in an individual that is specific for a self antigen called as autoantibodies. Autoantibodies can cause damage to cells and tissues and are produced in excess in systemic autoimmune diseases. The self-antigens, which are responsible for autoimmune disorders, were known as autogens.
Autoimmunity is a condition in which structural or functional damage is produced by the action of immunologically competent cells or antibodies against the normal components of the body. Autoimmunity literally means protection against self but it actually implies injury to self and therefore it has been criticized as a contradiction in terms. Autoallergy has been suggested as an acceptable alternative but the term autoimmunity has the sanction of wide usage.
The earliest example of autoimmunity was the observation by Metalnikoff (1900) that guinea pigs injected with their own spermatozoa produced sperm immobilizing antibodies. Donath and Landsteriner (1904) identified circulating autoantibodies in paroxysmal cold hemoglobinuria – a hemolysin which binds with the patient’s erythrocytes at low temperatures and produces complement dependent hemolysis on warming. This was the first description of an autoimmune disease in human beings. Dameshek and Schwarts (1938) established the autoimmune basis of acute hemolytic anemia.
The following criteria have to be satisfied before establishing the autoimmune etiology of disease:
1. An autoimmune response, humoral, cellular or both, must be regularly associated with the disease.
2. The antigen responsible for the immune response must be identified, isolated and characterized.
3. The same antigen must induce in experimental animals immunopathological changes as in the disease.
4. Passive transfer of the disease must be possible by transfer of antibodies or sensitized lymphocytes.
Diseases of autoimmune origin usually exhibit the following features:
i) An elevated level of immunoglobulins
ii) Demonstrable autoantibodies
iii) Deposition of immunoglobulins or their derivatives at sites of election, such renal glomeruli.
iv) Accumulation of lymphocytes and plasma cells at the sites of lesions.
v) Temporary or lasting benefit from corticosteroid or other immunosuppressive therapy.
vi) The occurrence of more than one type of autoimmune lesion in an individual.
vii) A genetic predisposition towards autoimmunity.
MECHANISM OF AUTOIMMUNITY:
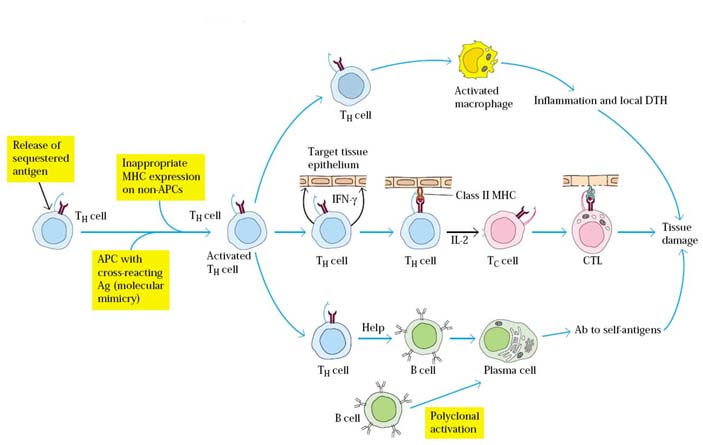
A variety of mechanisms have been proposed to account for the T-cell mediated generation of autoimmune diseases namely release of sequestered antigens, molecular mimicry, inappropriate expression of MHC molecules and polyclonal B-cell activation.
Release of Sequestered Antigens:
Hidden or sequestered antigens may not be recognized as self-antigens. When such antigens are released into circulation, they may induce an immune response. A few tissue antigens are known to fall in this category. For example, sperm arise late in development and are sequestered from the circulation. However, after a vasectomy, some sperm antigens are released into the circulation and can induce autoantibody formation in some men and cause autoimmunity against sperms. Similarly, the release of lens protein after eye damage or of heart-muscle antigens after myocardial infarction has been shown to lead to autoantibody formation on occasion.
Molecular Mimicry:
A number of viruses and bacteria have been shown to possess antigenic determinants that are identical or similar to normal host cell components which were referred as molecular mimicry. Such molecular mimicry appears in a wide variety of organisms. In one study, 600 different monoclonal antibodies specific for 11 different viruses were tested to evaluate their reactivity with normal tissue antigens. More than 3% of the virus-specific antibodies tested also bound to normal tissue, suggesting that molecular mimicry is a fairly common phenomenon.
Molecular mimicry has been suggested as one mechanism that leads to autoimmunity. One of the best examples of this type of autoimmune reaction is post-rabies encephalitis, which used to develop in some individuals who had received the rabies vaccine. In the past, the rabies virus was grown in rabbit brain-cell cultures, and preparations of the vaccine included antigens derived from the rabbit brain cells. In a vaccinated person, these rabbit brain-cell antigens could induce formation of antibodies and activated T cells, which could cross-react with the recipient’s own brain cells, leading to encephalitis. Cross-reacting antibodies are also thought to be the cause of heart damage in rheumatic fever, which can sometimes develop after a Streptococcus infection. In this case, the antibodies are to streptococcal antigens, but they cross-react with the heart muscle.
Inappropriate MHC II molecule expression:
The pancreatic beta cells of individuals with insulin-dependent diabetes mellitus (IDDM) express high levels of both class I and class II MHC molecules, whereas healthy beta cells express lower levels of class I and do not express class II at all. Similarly, thyroid acinar cells from those with Graves’ disease have been shown to express class II MHC molecules on their membranes. This inappropriate expression of class II MHC molecules, which are normally expressed only on antigen presenting cells, may serve to sensitize TH cells to peptides derived from the beta cells or thyroid cells, allowing activation of B cells or TC cells or sensitization of TH1 cells against self-antigens.
Polyclonal B-cell Activation:
A number of viruses and bacteria can induce nonspecific polyclonal B-cell activation. Gram-negative bacteria, cytomegalovirus, and Epstein-Barr virus (EBV) are all known to be such polyclonal activators, inducing the proliferation of numerous clones of B cells that express IgM in the absence of TH cells. If B cells reactive to self-antigens are activated by this mechanism, auto-antibodies can appear. For instance, during infectious mononucleosis, which is caused by EBV, a variety of auto-antibodies are produced, including autoantibodies reactive to T and B cells, rheumatoid factors, and antinuclear antibodies. Similarly, lymphocytes from patients with SLE produce large quantities of IgM in culture, suggesting that they have been polyclonally activated. Many AIDS patients also show high levels of nonspecific antibody and auto-antibodies to RBCs and platelets. These patients are often coinfected with other viruses such as EBV and cytomegalovirus, which may induce the polyclonal B-cell activation that results in auto-antibody production which intern results in autoimmunity.
Few other mechanisms are as follows: Cells or tissues may undergo antigenic alteration as a result of physical, chemical or biological influences. Such altered or neoantigens may elicit an immune response. Breakdown of immunological homeostasis may lead to cessation of tolerance and the emergence of forbidden clones of immunocompletent cells capable of mounting immune response against self-antigens. A variety of T and B cell defects have been suggested as possible mechanisms of autoimmunity.
CLASSIFICATION OF AUTOIMMUNE DISEASES:
Based on the site of involvement and nature of lesions, autoimmune diseases may be classified as hemocytolytic, localized or organ specific, systemic or non-organ specific and transitory diseases.
I. HEMOLYTIC AUTOIMMUNE DISEASES:
1. AUTOIMMUNE HEMOLYTIC ANEMIAS:
An individual with autoimmune hemolytic anemia makes auto-antibody to RBC antigens, triggering complement mediated lysis or antibody-mediated Opsonization and phagocytosis of the red blood cells. One form of autoimmune anemia is drug-induced: when certain drugs such as penicillin or the anti-hypertensive agent methyldopa interact with red blood cells, the cells become antigenic. The immunodiagnostic test for autoimmune hemolytic anemias generally involves a Coombs test, in which the red cells are incubated with an anti–human IgG antiserum. If IgG auto-antibodies are present on the red cells, the cells are agglutinated by the antiserum.
Autoantibodies against erythrocytes are two groups’ namely cold and warm autoantibodies. The cold autoantibodies are generally, complete agglutinating antibodies belonging to the IgM class and agglutinate erythrocytes at 4*C but not at 37*C. Cold agglutinins were first detected by Donath and Landsteiner in paroxysmal cold hemoglobinuria. This condition, which used to frequently accompany syphilitic infection, is seldom seen nowadays. Cold agglutinins are also seen in primary atypical pneumonia, trypanosomiasis and black water fever. The warm autoantibodies are generally incomplete, nonagglutinating antibodies belonging usually to the IgG class. They can be shown coating ghe erythrocytes in the direct coombs test. Warm antierythrocyte antibodies are frequently seen in patients taking certain drugs such as sulphonamides, antibiotics and alpha methyl dopa.
2. AUTOIMMUNE THROMBOCYTOPENIA:
Autoantibodies directed against platelets occur in idiopathic thrombocytopenic purpura. Sedormid purpura is an instance of immune response against drug induced neoantigens on platelets. This condition is traditionally considered antibody mediated hypersensitivity.
3. AUTOIMMUNE LEUCOPENIA:
Nonagglutinating antileucocyte antibodies can be demonstrated in serum of patients with systemic lupus erythermatosus and rheumatoid arthritis.
II. LOCALIZED OR ORGAN SPECIFIC AUTOIMMUNE DISEASES:
In an organ-specific autoimmune disease, the immune response is directed to a target antigen unique to a single organ or gland, so that the manifestations are largely limited to that organ. The cells of the target organs may be damaged directly by humoral or cell-mediated effector mechanisms. Alternatively, the antibodies may over stimulate or block the normal function of the target organ.
A. DIRECT CELLULAR DAMAGE:
Autoimmune diseases involving direct cellular damage occur when lymphocytes or antibodies bind to cell-membrane antigens, causing cellular lysis and/or an inflammatory response in the affected organ. Gradually, the damaged cellular structure is replaced by connective tissue (scar tissue), and the function of the organ declines. This section briefly describes a few examples of this type of autoimmune disease.
1. HASHIMOTO’S THYROIDITIS:
In Hashimoto’s thyroiditis, which is most frequently seen in middle-aged women, an individual produces auto-antibodies and sensitized TH1 cells specific for thyroid antigens. The DTH response is characterized by an intense infiltration of the thyroid gland by lymphocytes, macrophages, and plasma cells, which form lymphocytic follicles and germinal centers. The ensuing inflammatory response causes a goiter, or visible enlargement of the thyroid gland, a physiological response to hypothyroidism. Antibodies are formed to a number of thyroid proteins, including thyroglobulin and thyroid peroxidase, both of which are involved in the uptake of iodine. Binding of the auto-antibodies to these proteins interferes with iodine uptake and leads to decreased production of thyroid hormones (hypothyroidism).
2. PERNICIOUS ANEMIA:
Pernicious anemia is caused by auto-antibodies to intrinsic factor, a membrane-bound intestinal protein on gastric parietal cells. Intrinsic factor facilitates uptake of vitamin B12 from the small intestine. Binding of the auto-antibody to intrinsic factor blocks the intrinsic factor–mediated absorption of vitamin B12. In the absence of sufficient vitamin B12, which is necessary for proper hematopoiesis, the number of functional mature red blood cells decreases below normal. Pernicious anemia is treated with injections of vitamin B12, thus circumventing the defect in its absorption.
3. GOODPASTURE’S SYNDROME:
In Goodpasture’s syndrome, auto-antibodies specific for certain basement-membrane antigens bind to the basement membranes of the kidney glomeruli and the alveoli of the lungs. Subsequent complement activation leads to direct cellular damage and an ensuing inflammatory response mediated by a buildup of complement split products. Damage to the glomerular and alveolar basement membranes leads to progressive kidney damage and pulmonary hemorrhage. Death may ensue within several months of the onset of symptoms. Biopsies from patients with Goodpasture’s syndrome stained with fluorescent-labeled anti-IgG and anti-C3b reveal linear deposits of IgG and C3b along the basement Membranes.
4. INSULIN DEENDENT DIABETES MELLITUS:
A disease afflicting 0.2% of the population, insulin-dependent diabetes mellitus (IDDM) is caused by an autoimmune attack on the pancreas. The attack is directed against specialized insulin-producing cells (beta cells) that are located in spherical clusters, called the islets of Langerhans, scattered throughout the pancreas. The autoimmune attack destroys beta cells, resulting in decreased production of insulin and consequently increased levels of blood glucose. Several factors are important in the destruction of beta cells. First, activated CTLs migrate into an islet and begin to attack the insulin producing cells. Local cytokine production during this response includes IFN-g, TNF-b, and IL-1. Auto-antibody production can also be a contributing factor in IDDM. The first CTL infiltration and activation of macrophages, frequently referred to as insulitis, is followed by cytokine release and the presence of auto-antibodies, which leads to a cell-mediated DTH response. The subsequent beta-cell destruction is thought to be mediated by cytokines released during the DTH response and by lytic enzymes released from the activated macrophages. Auto-antibodies to beta cells may contribute to cell destruction by facilitating either antibody-plus-complement lysis or antibody-dependent cell-mediated cytotoxicity (ADCC).
5. ADDISON’S DISEASE:
The immunological basis of Addison’s disease is suggested by lymphocytic infilatration of the adrenal glands and the presence of circulating antibodies directed against the cells of the zona glomerulasa. Similar lesions can be produced in experimental animals by immunization with adrenal tissue in Freund’s adjuvant.
6. AUTOIMMUNE ORCHITIS:
Experimental allergic orchitis with progressive damage to germinal epithelium ad aspermatogenesis can be induced in guinea pigs by the injection of autogenous or allogenic testes with Freund’s adjuvant. A similar condition sometimes follows mumps orchitis. Lymphocytic infiltration of the testes and circulating antibodies to the sperms and germinal cells can be demonstrated in this condition.
7. AUTOIMMUNE DISEASEE OF THE EYE:
Two types of autoimmune diseases are seen in the eye. Cataract surgery sometimes leads to intraocular inflammation caused by the autoimmune response to the lens proteins. This is known as phacoanaphylaxis. Perforating injuries of the eye, particularly those involving the iris or ciliary bodies are often followed by sympathetic ophthalmia in the opposite eye. The disease can be produced in experimental animals by immunization with uveal or retinal tissue in Freund’s adjuvant and can be passively transferred with the spleen or lymph node cells but not with serum.
8. AUTOIMMUNE DISEASEE OF THE NERVOUS SYSTEM:
The neuroparalytic accidents following rabies vaccination represent injury to the nervous system by the immune response against the sheep nervous tissue in the vaccine which cross reacts with human nerve tissue. An essentially similar condition, experimental allergic encephalomyelitis (EAE) can be produced in animals by immunization with nervous tissue in Freund’s adjuvant. The encephalogenic protein has been identified as the myelin basic protein (MBP) which shows no species specificity.
Idiopathic polyneuritis (Guillain-Barre syndrome) is considered to be an autoimmune response against the peripheral nervous tissue. It can be reproduced in experimental animals by immunization with peripheral nervous tissue in an adjuvant.
9. AUTOIMMUNE DISEASE OF SKIN:
Three serious diseases of the skin are considered to have an autoimmune basis. Pemphigus vulgaris may be caused by an antibody to the intracellular cement substance. In bullous pemphigoid, antibodies directed against the dermal epithelial junction have been demonstrated. Specific antibodies in dermatitis herpetiformis have not been identified.
B. BLOCKING ANTIBODIES OR STIMULATING ANTIBODIES:
`In some autoimmune diseases, antibodies act as agonists, binding to hormone receptors in lieu of the normal ligand and stimulating inappropriate activity. This usually leads to an overproduction of mediators or an increase in cell growth. Conversely, auto-antibodies may act as antagonists, binding hormone receptors but blocking receptor function. This generally causes impaired secretion of mediators and gradual atrophy of the affected organ.
1. GRAVE’S DISEASE:
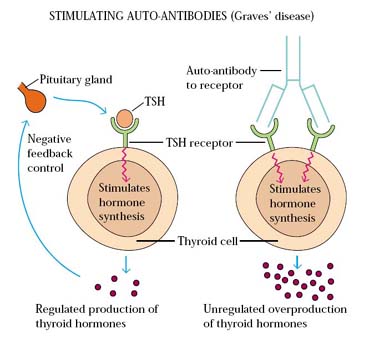
The production of thyroid hormones is carefully regulated by thyroid-stimulating hormone (TSH), which is produced by the pituitary gland. Binding of TSH to a receptor on thyroid cells activates adenylate cyclase and stimulates the synthesis of two thyroid hormones, thyroxine and triiodothyronine. A patient with Graves’ disease produces auto-antibodies that bind the receptor for TSH and mimic the normal action of TSH, activating adenylate cyclase and resulting in production of the thyroid hormones. Unlike TSH, however, the autoantibodies are not regulated, and consequently they over stimulate the thyroid. For this reason these auto-antibodies are called long-acting thyroid-stimulating (LATS) antibodies.
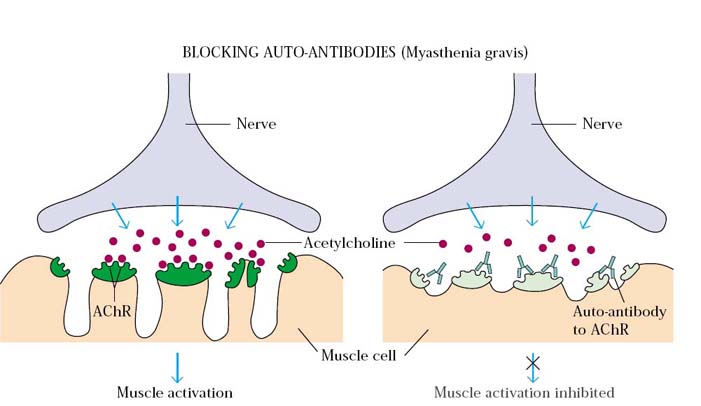
2. MYASTHENIA GRAVIS:
Myasthenia gravis is the prototype autoimmune disease mediated by blocking antibodies. A patient with this disease produces auto-antibodies that bind the acetylcholine receptors on the motor end-plates of muscles, blocking the normal binding of acetylcholine and also inducing complement mediated lysis of the cells. The result is a progressive weakening of the skeletal muscles. Ultimately, the antibodies destroy the cells bearing the receptors. The early signs of this disease include drooping eyelids and inability to retract the corners of the mouth, which gives the appearance of snarling. Without treatment, progressive weakening of the muscles can lead to severe impairment of eating as well as problems with movement. However, with appropriate treatment, this disease can be managed quite well and afflicted individuals can lead a normal life.
III. SYSTEMIC OR NON-ORGAN SPECIFIC AUTOIMMUNE DISEASE:
This group includes conditions characterized by immune response against variety of self-antigens and damage to several organs and tissue systems.
1. SYSTEMIC LUPUS ERYTHEMATOSUS:
One of the best examples of a systemic autoimmune disease is systemic lupus erythermatosus (SLE), which typically appears in women between 20 and 40 years of age; the ratio of female to male patients is 10:1. SLE is characterized by fever, weakness, arthritis, skin rashes, pleurisy, and kidney dysfunction. Lupus is more frequent in African-American and Hispanic women than in Caucasians, although it is not known why this is so. Affected individuals may produce autoantibodies to a vast array of tissue antigens, such as DNA, histones, RBCs, platelets, leukocytes, and clotting factors; interaction of these auto-antibodies with their specific antigens produces various symptoms. Auto-antibody specific for RBCs and platelets, for example, can lead to complement-mediated lysis, resulting in hemolytic anemia and thrombocytopenia, respectively. When immune complexes of auto-antibodies with various nuclear antigens are deposited along the walls of small blood vessels, a type III hypersensitive reaction develops. The complexes activate the complement system and generate membrane-attack complexes and complement split products that damage the wall of the blood vessel, resulting in vasculitis and glomerulonephritis. Excessive complement activation in patients with severe SLE produces elevated serum levels of the complement split products C3a and C5a, which may be three to four times higher than normal. C5a induces increased expression of the type 3 complement receptor (CR3) on neutrophils, facilitating neutrophil aggregation and attachment to the vascular endothelium. As neutrophils attach to small blood vessels, the number of circulating neutrophils declines (neutropenia) and various occlusions of the small blood vessels develop (vasculitis). These occlusions can lead to widespread tissue damage. Laboratory diagnosis of SLE focuses on the characteristic antinuclear antibodies, which are directed against double stranded or single-stranded DNA, nucleoprotein, histones, and nucleolar RNA. Indirect immunofluorescent staining with serum from SLE patients produces various characteristic nucleus-staining patterns.
2. MULTIPLE SCLEROSIS:
Multiple sclerosis (MS) is the most common cause of neurological disability associated with disease in Western countries. The symptoms may be mild, such as numbness in the limbs, or severe, such as paralysis or loss of vision. Most people with MS are diagnosed between the ages of 20 and 40. Individuals with this disease produce autoreactive T cells that participate in the formation of inflammatory lesions along the myelin sheath of nerve fibers. The cerebrospinal fluid of patients with active MS contains activated T lymphocytes, which infiltrate the brain tissue and cause characteristic inflammatory lesions, destroying the myelin. Since myelin functions to insulate the nerve fibers, a breakdown in the myelin sheath leads to numerous neurologic dysfunctions. The cause of MS, like most autoimmune diseases, is not well understood. However, there are some suggestions that infection by certain viruses may predispose a person to MS. Certainly some viruses can cause demyelinating diseases, and it is tempting to speculate that virus infection plays a significant role in MS, but at present there is no definitive data implicating a particular virus.
3. RHEUMATOID ARTHRITIS:
Rheumatoid arthritis is a common autoimmune disorder, most often affecting women from 40 to 60 years old. The major symptom is chronic inflammation of the joints, although the hematologic, cardiovascular, and respiratory systems are also frequently affected. Many individuals with rheumatoid arthritis produce a group of auto-antibodies called rheumatoid factors that are reactive with determinants in the Fc region of IgG. The classic rheumatoid factor is an IgM antibody with that reactivity. Such auto-antibodies bind to normal circulating IgG, forming IgM-IgG complexes that are deposited in the joints. These immune complexes can activate the complement cascade, resulting in a type III hypersensitive reaction, which leads to chronic inflammation of the joints.
RF is detected by agglutination tests using as antigens particles coated with globulins. In Rose Waaler test, the original technique for detection of RF, sheep erythrocytes coated with a subagglutinating dose of antierythrocyte antibody (amboceptor) are used as the antigen in an agglutination test. In modifications of the test, latex and bentonite are used as the carrier particles for IgG. Antinuclear antibodies are frequently found in rheumatoid arthritis.
4. SJOGREN’S SYNDROME:
This is a triad of conjunctivitis sicca, dryness of the mouth, with or without salivary gland enlargement, and rheumatoid arthritis. The syndrome may occur in association with other collagen disease. Antinuclear antibodies and rheumatoid factor commonly occur in sera.
5.POLYARTERITIS NODOSA:
This is a necrotizing angitis involving medium sized arteries, ending fatally due to coronary thrombosis, cerebral hemorrhage or gastrointestinal bleeding. Polyarteritis is seen as a component of serum sickness and other toxic complex diseases. Immune complexes of hepatitis B virus antigen (HbsAg) in affected tissues, including the kidneys, have been demonstrated in 30-40 percent of patients. Though it has been suggested that Polyarteritis nodosa may be an autoimmune disease, the autoantibody responsible has not been identified.
IV. TRNSITORY AUTOIMMUNE DISEASES:
These include conditions such as anemia, thrombocytopenia or nephritis that follow certain infections or drug therapy. The infecting agent or drug induces antigenic alteration in some self-antigens. The immune response set up causes tissue damage. The disease is transient and undergoes spontaneous cure when the infection is controlled or the drug withdrawn.
PATHOGENESIS OF AUTOIMMUNITY:
Many diseases are considered to be autoimmune origin, based on their association with cellular or humoral immune responses against self-antigens. Autoantibodies are more easily detected then cellular autosensitisation. However, the mere presence of autoantibodies during the course of a disease does not prove their eitiological role. Autoantibody formation may be a result of tissue injury and the antibody may help in promoting immune elimination of the damaged cell or tissue elements. A typical example is lepromatous leprosy in which large amounts of autoantibodies are regularly found. It has been said that but for the lepra bacillus, lepromatous leprosy may have bee proposed as an autoimmune disease.
The relative importance of humoral and cellular immune processes in the etiology of autoimmune disease is not known. Antibodies may cause damage by the cytolytic or cytotoxic (i.e type 2 hypersensitivity mechanism) and toxic complex (i.e type 3 mechanisms) reactions. They are obviously important in hemocytolytic autoimmune diseases. A third mechanism of autoimmune tissue damage is by sensitized T lymphocytes (i.e type 4 hypersensitivity). It is likely that humoral and cellular immune responses may act synergistically in the production of some autoimmune diseases. For example experimental orchitis can be induced only when both types of immune responses are operative. Once initiated, most autoimmune responses tend to be self perpetuating. Their progress can be arrested by immunosuppressive therapy, though the degree of response to such therapy varies in different diseases.
TREATMENT:
Often, in organ-specific autoimmune disorders, the symptoms can be corrected by metabolic control. For example, hypothyroidism can be controlled by administration of thyroxine and thyrotoxicosis by antithyroid drugs. In pernicious anaemia, metabolic correction is achieved by injection of vitamin B12 and in myasthenia gravis by administration of cholinesterase inhibitors. When functions is lost and cannot be substituted by hormones, as may occur in lupus nephritis or chronic rheumatoid arthritis, tissue grafts or mechanical substitutes may be appropriate. In the case of tissue graft, protection from the immunological processes which necessitated the transplant may be required.
Conventional immunosuppressive therapy with antimitotic drugs can be used to damp down the immune response but because of the dangers involved, tends to be used only in life threatening disorders such as SLE and dermatomyositis. The potential of cyclosporine and related drugs has yet to be fully realized but quite dramatic results have been reported in the treatment of type I diabetes mellitus. Anti-inflammatory drugs are, of course, prescribed for rheumatoid diseases.
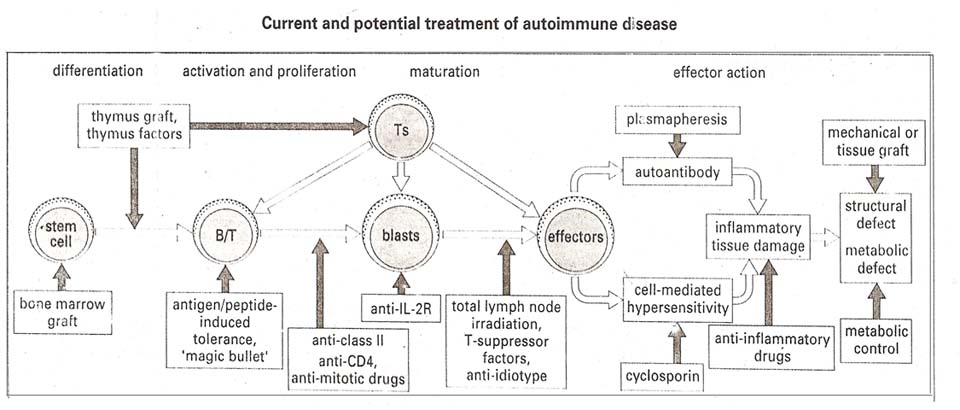
In particular, some experimental autoimmune diseases have been treated successfully with autoantigenic peptides and their analogues and by vaccination with autoreactive T-cells. This suggested that stimulating normally suppressive functions including the idiotype network could be promising.