ANTIGEN-ANTIBODY REACTIONS
Antigen-Antibody reactions refer to the interaction between antigens and antibodies. These are reactions are studied under two different headings namely in vivo and in vitro reactions.
In vivo Ag-Ab reactions:
These reactions refer to the Ag-Ab reactions occurring inside the host. In the body, they form the basis of antibody mediated immunity in infectious diseases or of tissue injury in some types of hypersensitivity and autoimmune diseases. Functions of antibodies are also included under this category. They are
a. Agglutination
b. Neutralization
c. Opsonization (IgG1 and 3)
d. Complement fixation (IgM and IgG)
e. Induction of inflammation (IgE)
(Explain in detail from immunoglobulin notes)
In vitro Ag-Ab reactions:
These reactions refer to the Ag-Ab reactions occurring outside host. In the laboratory, they help in the diagnosis of infections, in epidemiological surveys, in the identification of infectious agents and of noninfectious antigens such as enzymes. Antigen-antibody reactions in vitro are known as serological reactions. All immunological techniques studied under this heading are of in vitro type.
The reactions between antigens and antibodies occur in three stages. The primary stage is the initial interaction between the two, without any visible effects. This reaction is rapid, occurs even at low temperatures and obeys the general laws of physical chemistry and thermodynamics. The reaction is reversible, the combination between antigen and antibody molecules being effected by the weaker intermolecular forces such as Van der Waal’s forces, ionic bonds and hydrogen bonding, rather than by the firmer covalent bonding. The primary reaction can be detected by estimating free and bound antigen or antibody separately in the reaction mixture by a number of physical and chemical methods, including the use of markers such as radioactive isotopes, fluorescent dyes or ferritin.
In most instances, but not all, the primary stage is followed by the secondary stage leading to demonstrable events such as precipitation, agglutination, lysis of cells, killing of live antigens, neutralization of toxins and other biologically active antigens, fixation of complement, immobilization of motile organisms and enhancement of phagocytosis. When such reactions were discovered one by one, it was believed that a different type of antibody was responsible for each type of reaction and the antibodies came to be designated by the reactions they were thought to produce. Thus, the antibody causing agglutination was called agglutinin, that causing precipitation precipitin, etc., and the corresponding antigen, agglutinogen, precipitinogen, etc., By the 1920s, this view was replaced by Zinsser’s Unitarian hypothesis which held that an antigen gave rise to only one antibody, which was capable of producing all the different reactions depending on the nature of the antigen and the conditions of the reaction. Both these extreme views are fallacious. While it is true that a single antibody can cause precipitation, agglutination and most of the other serological reactions, it is also true that an antigen can stimulate the production of different classes of immunoglobulin which differ in their reaction capacities as well as in other properties.
Some antigen-antibody reactions occurring in vivo initiate chain reactions that lead to neutralization of destruction of injury antigens or to tissue damage. These are the tertiary reactions and include humoral immunity against infectious disease as well as clinical allergy and other immunological disease.
GENERAL FEATURES:
Antigen-antibody reactions have the following general characteristics:
1. The reaction is specific, an antigen combining only with its homologous antibody and vice versa. The specificity, however, is not absolute and cross reactions may occur due to antigenic similarity or relatedness. The phenomenon was known as cross reactivity.
2. Entire molecules react and not fragment. When an antigenic determinants present in a large molecule or on a carrier particle reacts with its antibody, whole molecules or particles are agglutinated.
3. There is no denaturation of the antigen or the antibody during the reaction.
4. The combination occurs at the surface. Therefore, it is the surface antigens that are immunologically relevant. Antibodies to the surface antigens of infectious agents are generally protective.
5. The combination is firm but reversible. The firmness of the union is influenced by the affinity and avidity of the reaction. Affinity refers to the intensity of attraction between single epitope of the antigen and paratope of antibody molecules. It is a function of the closeness of fit between an epitope and the antigen combining region of antibody. Avidity is the strength of the bond after the formation of the antigen antibody complexes. It reflects the overall combining property of the various antibody molecules in an antiserum, possessing different affinity constants with the multiple epitopes of the antigen. Generally IgG possess greater affinity and IgM possess higher avidity and vice versa.
6. Both antigen and antibodies participate in the formation of agglutinates or precipitates.
7. Antigens and antibodies can combine in varying proportions, unlike chemicals with fixed valencies. Both antigens and antibodies are multivalent. Antibodies are generally bivalent, though IgM molecules may have five or ten combining sites. Antigens may have valencies up to hundreds.
STRENGTH OF Ag-Ab REACTIONS:
Low-affinity antibodies bind antigen weakly and tend to dissociate readily, whereas high-affinity antibodies bind antigen more tightly and remain bound longer. The association between binding sites on an antibody (Ab) with a monovalent antigen (Ag) can be described by the equation
Where k1 is the forward (association) rate constant and k_1 is the reverse (dissociation) rate constant. The ratio k1/k_1 is the association constant Ka (i.e., k1/k_1 = Ka), a measure of affinity. Because Ka is the equilibrium constant for the above reaction, it can be calculated from the ratio of the molar concentration of bound Ag-Ab complex to the molar concentrations of unbound antigen and antibody at equilibrium as follows:
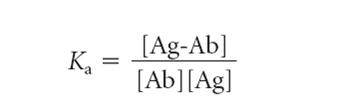
The affinity constant, Ka, can be determined by equilibrium dialysis or by various newer methods. Because equilibrium dialysis remains for many the standard against which other methods are evaluated, it is described here. This procedure uses a dialysis chamber containing two equal compartments separated by a semipermeable membrane. Antibody is placed in one compartment, and a radioactively labeled ligand that is small enough to pass through the semipermeable membrane is placed in the other compartment. Suitable ligands include haptens, oligosaccharides, and oligopeptides. In the absence of antibody, ligand added to compartment B will equilibrate on both sides of the membrane. In the presence of antibody, however, part of the labeled ligand will be bound to the antibody at equilibrium, trapping the ligand on the antibody side of the vessel, whereas unbound ligand will be equally distributed in both compartments.

Thus the total concentration of ligand will be greater in the compartment containing antibody. The difference in the ligand concentration in the two compartments represents the concentration of ligand bound to the antibody (i.e., the concentration of Ag-Ab complex). The higher the affinity of the antibody, the more ligand is bound. Since the total concentration of antibody in the equilibrium dialysis chamber is known, the equilibrium equation can be rewritten as:
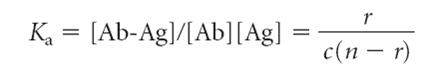
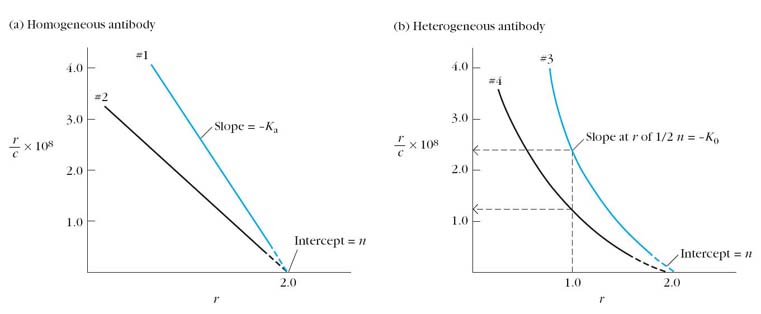
Where r equals the ratio of the concentration of bound ligand to total antibody concentration, c is the concentration of free ligand, and n is the number of binding sites per antibody molecule. This expression can be rearranged to give the Scatchard equation:
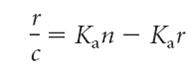
Values for r and c can be obtained by repeating the equilibrium dialysis with the same concentration of antibody but with different concentrations of ligand. If Ka is a constant, that is, if all the antibodies within the dialysis chamber have the same affinity for the ligand, then a Scatchard plot of r/c versus r will yield a straight line with a slope of Ka. As the concentration of unbound ligand c increases, r/c approaches 0, and r approaches n, the valency, equal to the number of binding sites per antibody molecule. Most antibody preparations are polyclonal, and Ka is therefore not a constant because a heterogeneous mixture of antibodies with a range of affinities is present. A Scatchard plot of heterogeneous antibody yields a curved line whose slope is constantly changing, reflecting this antibody heterogeneity. With this type of Scatchard plot, it is possible to determine the average affinity constant, K0, by determining the value of Ka when half of the antigen-binding sites are filled. This is conveniently done by determining the slope of the curve at the point where half of the antigen binding sites are filled.
MEASUREMENT OF ANTIGEN-ANTIBODY:
Many methods are available for the measurement of antigens and antibodies participating in the primary, secondary or tertiary reactions. Measurement may be in terms of mass or more commonly as units or titre. Antigens may also be titrated against sera. Two important parameters of serological tests are sensitivity and specificity. Sensitivity refers to the ability of the test to detect even very minute quantities of antigen or antibody. When a test is highly sensitive, false negative results will be absent or minimal. Specificity refers to the ability of the test to detect reactions between homologous antigens and antibodies only and with no other. In a highly specific test, false positive reactions will be absent or minimal. In general, sensitivity and specificity of a test will be in inverse proportion.
Techniques used to measure antigen-antibody called as immunotechniques, which broadly classified into two categories namely primary techniques and secondary techniques. Primary techniques utilized the physical changes occurring during antigen antibody interaction for estimation whereas secondary techniques utilize labels or markers to estimate antigen and antibodies.
1. PRIMARY IMMUNOTECHNIQUES:
Primary immunotechniques utilizes the physical changes like precipitation and agglutination occurring during antigen-antibody reaction for estimation.
1.1 PRECIPITATION REACTIONS:
The interaction between an antibody and a soluble antigen in aqueous solution forms a lattice that eventually develops a visible precipitation. Antibody that forms precipitation is known as precipitin. This process is called as precipitation reaction. Formation of an Ag-Ab lattice depends on the valency of both antigen and antibody. The antibody should be bivalent. The antigen must be either bivalent or polyvalent. Zone of equivalence is a point at which the maximum precipitation occurs. This reaction is widely used in several immunological techniques. Precipitation occurs in both fluid and gel.
1.1.1 PRECIPITATION IN FLUID:
A quantitative precipitation reaction can be performed by placing a constant amount of antibody in a series of tubes and adding increasing amounts of antigen to the tubes. At one time this method was used to measure the amount of antigen or antibody present in a sample of interest. After the precipitate forms, each tube is centrifuged to pellet the precipitate, the supernatant is poured off, and the amount of precipitate is measured. Plotting the amount of precipitate against increasing antigen concentrations yields a precipitin curve. Excess of either antibody or antigen interferes with maximal precipitation, which occurs in the so-called equivalence zone, within which the ratio of antibody to antigen is optimal. As a large multimolecular lattice is formed at equivalence, the complex increases in size and precipitates out of solution. In Precipitation reactions under conditions of antibody excess or antigen excess, extensive lattices do not form and precipitation is inhibited. Although the quantitative precipitation reaction is seldom used experimentally today, the principles of antigen excess, antibody excess, and equivalence apply to many Ag-Ab reactions.
The precipitation test may be carried out either as a qualitative or as a quantitative test. It is very sensitive for detecting antigens and little as 1mg of protein can be detected by precipitation tests. It therefore finds forensic application in the identification of blood and seminal stains and in testing for food adulterants. Precipitants are relatively less sensitive for the detection of antibodies.

Applications:
The following types of precipitation and flocculation tests are in common use:
Ring test: This, the simplest type of precipitation test, consists of layering the antigen solution over a column of antiserum in a narrow tube. A precipitate forms at the junction of the two liquids. Ring tests have only a few clinical applications now. Examples are Ascoli’s thermoprecipitin test and the grouping of streptococci by the Lancefield technique.
Slide test: When a drop, each of the antigen and antiserum are placed on a slide and mixed by shaking flocculates appear. The VDRL (Venereal Disease Research Laboratory) test for syphilis caused by Treponema Pallidum is an example of slide flocculation.
Tube test: The kahn test for syphilis is an example of a tube flocculation test. A quantitative tube flocculation test is employed for the standardization of toxins and toxoids. Serial dilutions of the toxin/toxoid are added to the tubes containing a fixed quantity of the antitoxin. The amount of toxin or toxoid that flocculates optimally with one unit of the antitoxin is defined as Lf dose.
1.1.2. PRECIPITATION IN GEL:
There are several advantages in allowing precipitation to occur in a gel rather than in a liquid medium. The reaction is visible as a distinct band of precipitation, which is stable and can be stained for preservation, if necessary. As each antigen-antibody reaction gives raise to a line of precipitation, the number of different antigens in the reacting mixture can be readily observed. Immunodiffusion also indicates identity, cross reaction and nonidentity between different antigens. Immunodiffusion is usually performed in a soft 1% agar gel. Immunodiffusion divided into two type namely simple immunodiffusion and electroimmunodiffusion.
I. SIMPLE IMMUNODIFFUSION:
Simple Immunodiffusion refers to the diffusion of antigens and antibodies without any external forces. This technique further divided into two types namely Single immunodiffusion and double immunodiffusion.
i) Single Immunodiffusion:
In this type of immunodiffusion, only antigen or antibody diffuses in gel.
1. Single diffusion in one dimension (Oudin Procedure)
The antibody is incorporated in agar gel in a test tube and the antigen solution is layered over it. The antigen diffuses downward through the agar gel, forming a line of precipitation that appears to move downwards. This is due to the precipitation formed at the advancing front of the antigen and is dissolved as the concentration of antigen at the site increases due to diffusion. The number of bands indicates the number of different antigens present.

2. Single diffusion in two dimensions (Radial immunodiffusion):
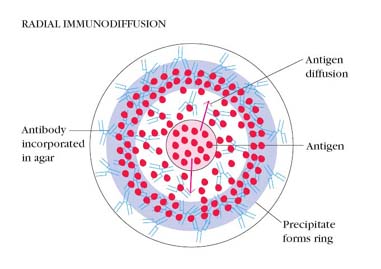
Here the antiserum is incorporated in agar gel poured on a flat surface of slide or Petri dish. The antigen is added to the wells cut on the surface of the gel. The antigen diffuses radially from the well and forms ring shaped bands of precipitation (halos) concentrically around the well. The diameter of the halo gives an estimate of the concentration of the antigen. This method has been employed for the estimation of the immunoglobulin classes in sera. It has also been used for screening sera for antibodies to influenza viruses. This method is also otherwise known as Mancini method.
The Mancini method is routinely used to quantitate serum levels of IgM, IgG and IgA by incorporating class-specific anti-isotype antibody into the agar. The technique is also applied to determine the concentrations of complement components in serum. The Mancini method cannot detect antigens present in concentrations below 5-10ug/ml; this moderate sensitivity is the major limitation of the radial immunodiffusion method. This method is used to determine alpha fetoprotein occurring in certain liver tumors.
ii) DOUBLE IMMUNODIFFUSION:
In double immunodiffusion, double refers to the diffusion of both antigens and antibodies. Under this heading, simple double immunodiffusion, Ouchterlony procedure and immunoelectrophoresis are studied.
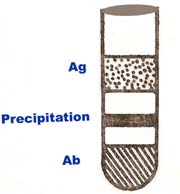
1. Simple double immunodiffusion ( Oakley Fulthorpe procedure):
It is double immunodiffusion in one dimension. Here, the antibody is incorporated in gel, above which is placed a column of plain agar. The antigen is layered on top of this. The antigen and antibody move towards each other through the intervening column of plain agar and form a band of precipitate where they meet at optimum proportion. This technique usually used to identify the presence of either antigen or antibody.
2. Ouchterlony Procedure:
It is double diffusion in two dimensions. This is the immunodiffusion method most widely employed and helps to compare different antigens and antisera directly. Agar gel is poured on a slide and wells are cut using a template. The antiserum is placed in the central well and different antigens in the surrounding well. If two adjacent antigens are identical, the lines of precipitate formed by them will fuse and prove “V” shaped curve. If they are unrelated, the lines will cross each other and provide “X” shaped curve. Cross reaction or partial identiy is indicated by spur formation and provide “Y” shaped curve. This method was a routine technique for the diagnosis of smallpox. When extracts of smallpox lesions are tested against the antiserum, precipitation lines can be seen within 2-6 hours. A special variety of double diffusion in two dimensions is the Elk test for toxigenicity in diphtheria bacilli. When diphtheria bacilli are streaked at right angles to a filter paper strip carrying the antitoxin implanted on a plate of suitable medium, arrowhead shaped lines of precipitation appear on incubation, if the bacillus is toxigenic.
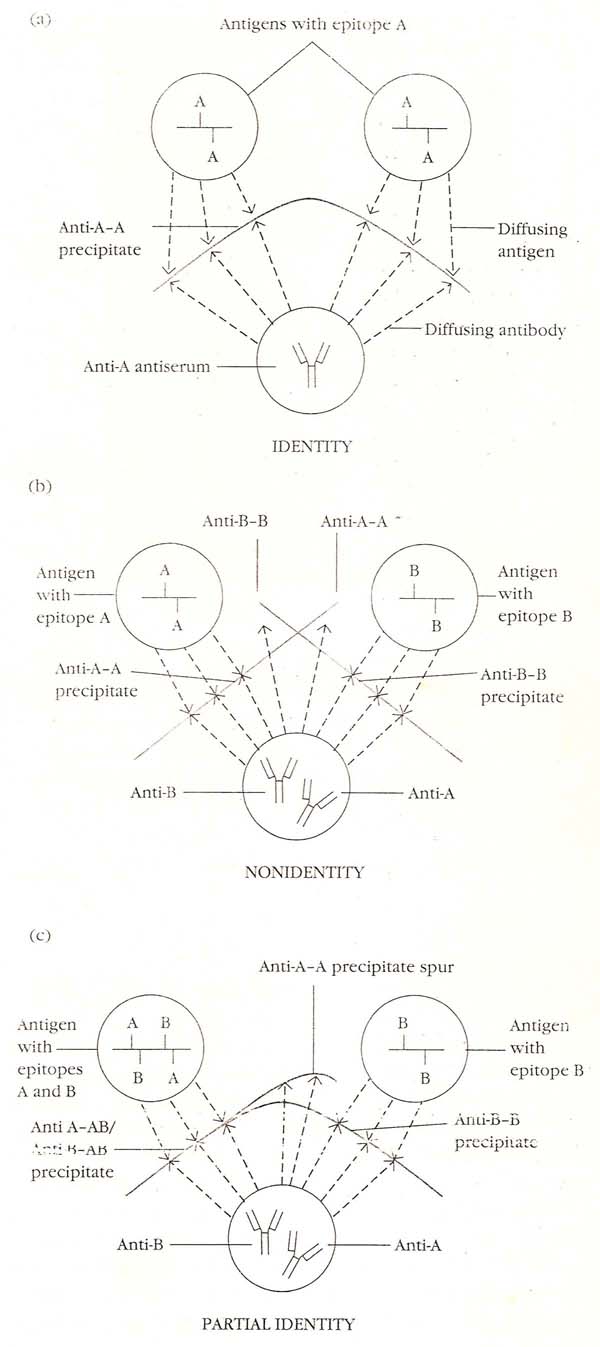
3. Immunoelectrophoresis:
Immunoelectrophoresis, the antigen mixture is first electrophoresed to separate its components by charge. Troughs are then cut into the agar gel parallel to the direction of the electric field, and antiserum is added to the troughs. Antibody and antigen then diffuse toward each other and produce lines of precipitation where they meet in appropriate proportions. Immunoelectrophoresis is used in clinical laboratories to detect the presence or absence of proteins in the serum. A sample of serum is electrophoresed, and the individual serum components are identified with antisera specific for a given protein or immunoglobulin class.
This technique is useful in determining whether a patient produces abnormally low amounts of one or more isotypes, characteristic of certain immunodeficiency diseases. It can also show whether a patient overproduces some serum protein, such as albumin, immunoglobulin, or transferrin. The immunoelectrophoretic pattern of serum from patients with multiple myeloma, for example, shows a heavy distorted arc caused by the large amount of myeloma protein, which is monoclonal Ig and therefore uniformly charged. Because immunoelectrophoresis is a strictly qualitative technique that only detects relatively high antibody concentrations (greater than several hundred ug/ml), it utility is limited to the detection of quantitative abnormalities only when the departure from normal is striking, as in immunodeficiency states and immunoproliferative disorders. Over 30 different proteins can be identified by this method in human serum. This is useful for testing normal and abnormal proteins in serum and urine. It is used for the identification of myeloma proteins.

II) ELECTROIMMUNODIFFUSION:
The development of precipitin lines can be speeded up by electrically driving the antigen and antibody. Various methods have been described combining electrophoresis with diffusion. In this technique, charge of antibody and antigen plays vital role. Of these, one dimensional double electroimmunodiffusion (Counter currnet Immunoelectrophoresis), one dimensional single electrodiffusion (rocket electrophoresis) and two dimensional immunoelectrophoresis are used frequently in the clinical laboratory.
1. Countercurrent Immunoelectrophoresis (CIE):
This involves simultaneous electrophoresis of the antigen and antibody in gel in opposite directions resulting in precipitation at a point between them. This method produces visible precipitation lines within thirty minutes and is ten times more sensitive than the standard double diffusion techniques. The clinical applications are for detecting various antigens such as alpha fetoprotein in serum and specific antigens of cryptococccus and meningococcus in cerebrospinal fluid.
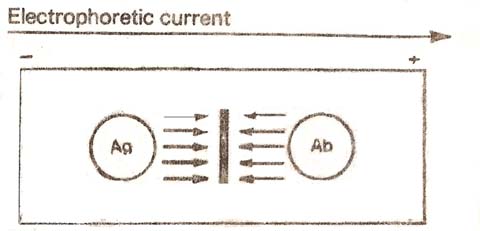
This method is used to determine anti-nuclear factor in systemic lupus erythromatosus. It is also used to determine hepatitis B antigen or antibody present in eh serum.
2. Rocket electrophoresis:
The antiserum to the antigen to be quantified is incorporated in agarose and gelled on the glass slide. The antigen, in increasing concentrations, is placed in wells punched in the set gel. The antigen is then electrophoresed into the antibody containing agarose. The pattern of immunoprecipitation resembles a rocket and hence the name. The main application of this technique is for quantitative estimation of antigens. This method is useful in the quantitative determination of proteins such as albumin, transferrin, etc.; one limitation of rocketelectrophoresis is the need for the antigen to be negatively charged for electrophoretic movement within the agar matrix.

3. Two dimensional gel electrophoresis:
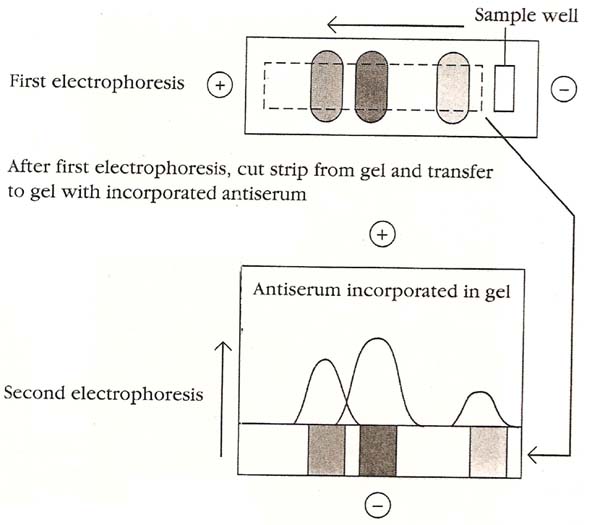
Several antigens in a complex mixture can be quantitated simultaneously with a modification of rocket electrophoresis called two-dimensional immunoelectrophoresis. In this technique antigen is first separated into components by electrophoresis. The gel is then laid over another agar gel containing antiserum and electrophoresis is repeated at right angles to the first direction, forming precipitin peaks similar to those obtained with rocket electrophoresis. Measurement of the size of the peaks allows quantification of a number of proteins in a complex antigen mixture.
1.2. AGGLUTINATIONS:
The interaction between antibody and a particulate antigen results in visible clumping called agglutination. Antibodies that produce such reactions are called agglutinins. Agglutination reactions are similar in principle to precipitation reactions; they depend on the cross linking of polyvalent antigens. Just as an excess of antibody inhibits precipitation reactions, such excess can also inhibit agglutination reactions; this inhibition is called the prozone effect. Because prozone effects can be encountered in many types of immunoassays, understanding the basis of this phenomenon is of general importance. Several mechanisms can cause the prozone effect. First, at high antibody concentrations, the number of antibody binding sites may greatly exceed the number of epitopes. As a result, most antibodies bind antigen only univalently instead of multivalently. Antibodies that bind univalently cannot crosslink one antigen to another. Prozone effects are readily diagnosed by performing the assay at a variety of antibody (or antigen) concentrations. As one dilutes to an optimum antibody concentration, one sees higher levels of agglutination or whatever parameter is measured in the assay being used. When one is using polyclonal antibodies, the prozone effect can also occur for another reason. The antiserum may contain high concentrations of antibodies that bind to the antigen but do not induce agglutination; these antibodies, called incomplete antibodies, are often of the IgG class. At high concentrations of IgG, incomplete antibodies may occupy most of the antigenic sites, thus blocking access by IgM, which is a good agglutinin. This effect is not seen with agglutinating monoclonal antibodies. The lack of agglutinating activity of an incomplete antibody may be due to restricted flexibility in the hinge region, making it difficult for the antibody to assume the required angle for optimal cross-linking of epitopes on two or more particulate antigens. Alternatively, the density of epitope distribution or the location of some epitopes in deep pockets of a particulate antigen may make it difficult for the antibodies specific for these epitopes to agglutinate certain particulate antigens. When feasible, the solution to both of these problems is to try different antibodies that may react with other epitopes of the antigen that do not present these limitations.
HAEMAGGLUTINATION (SLIDE AGGLUTINATION):
Agglutination reactions are routinely performed to type red blood cells (RBCs). In typing for the ABO antigens, RBCs are mixed on a slide with antisera to the A or B blood-group antigens. If the antigen is present on the cells, they agglutinate, forming a visible clump on the slide. Determination of which antigens are present on donor and recipient RBCs is the basis for matching blood types for transfusions. At neutral pH, red blood cells are surrounded by a negative ion cloud that makes the cells repel one another this repulsive force is called zeta potential. Because of its size and pentameric in nature, IgM can overcome the zeta potential and cross link red blood cells, leading to agglutination. The smaller size and bivalency of IgG makes it less able to overcome the zeta potential. For this reason, IgM is more effective than IgG in agglutinating red blood cells.
BACTERIAL AGGLUTINATION (TUBE AGGLUTINATION):
A bacterial infection often elicits the production of serum antibodies specific for surface antigens on the bacterial cells. The presence of such antibodies can be detected by bacterial agglutination reactions. Serum from a patient thought to be infected with a given bacterium is serially diluted in an array of tubes to which the bacteria is added. The last tube showing visible agglutination will reflect the serum antibody titer of the patient. The agglutinin titer is defined as the reciprocal of the greatest serum dilution that elicits a positive agglutination reaction. For example, if serial twofold dilutions of serum are prepared and if the dilution of 1/640 shows agglutination but the dilution of 1/1280 does not, then the agglutination titer of the patient’s serum is 640. In some cases serum can be diluted up to 1/50,000 and still show agglutination of bacteria. The agglutinin titer of an antiserum can be used to diagnose a bacterial infection. Patients with typhoid fever, for example, show a significant rise in the agglutination titer to Salmonella typhi. Agglutination reactions also provide a way to type bacteria. For instance, different species of the bacterium Salmonella can be distinguished by agglutination reactions with a panel of typing antisera.
Widal test is used for the diagnosis of typhoid fever. In typhoid patients, the serum contains antibodies to salmonella. In Widal test two antigens are used. They are antigen H and the flagellar antigen and O antigen, the somatic antigen. When antiserum of the patients is added to the antigens, the antigens are clumped and identified. The Weil Felix reaction for serodiagnosis of typhus fever is a heterophile agglutination test and is based on the sharing of a common antigen between typhus rickettsiae and some strains of proteus bacilli. Another example of the heterophile agglutination test is the streptococcus MG agglutination test for the diagnosis of primary atypical pneumonia. Brucella agglutination test is employed for brucellosis.
PASSIVE AGGLUTINATION:
The sensitivity and simplicity of agglutination reactions can be extended to soluble antigens by the technique of passive hemagglutination. In this technique, antigen-coated red blood cells are prepared by mixing a soluble antigen with red blood cells that have been treated with tannic acid or chromium chloride, both of which promote adsorption of the antigen to the surface of the cells. Serum containing antibody is serially diluted into microtiter plate wells, and the antigen-coated red blood cells are then added to each well; agglutination is assessed by the size of the characteristic spread pattern of agglutinated red blood cells on the bottom of the well, like the pattern seen in agglutination reactions.
Over the past several years, there has been a shift away from red blood cells to synthetic particles, such as latex beads, as matrices for agglutination reactions. Once the antigen has been coupled to the latex beads, the preparation can either be used immediately or stored for later use. The use of synthetic beads offers the advantages of consistency, uniformity, and stability. Furthermore, agglutination reactions employing synthetic beads can be read rapidly, often within 3 to 5 minutes of mixing the beads with the test sample. Whether based on red blood cells or the more convenient and versatile synthetic beads, agglutination reactions are simple to perform, do not require expensive equipment, and can detect small amounts of antibody (concentrations as low as nanograms per milliliter). Latex agglutination test (Latex fixation test) are widely employed in the clinical laboratory for the detection of hepatitis B, ASO, CRP, RA factor, HCG and many other antigens.
A special type of passive hemagglutination test is the Rose-Waaler test. In rheumatoid arthritis, an autoantibody (RA factor) appears in the serum, which acts as an antibody to gammaglobulin. The RA factor is able to agglutinate red cells coated with globulins. The antigen used for the test is a suspension of sheep erythrocytes sensitized with a subagglutinating dose of rabbit antisheep erythrocyte antibody (amboceptor).
AGGLUTINATION INHIBITION:
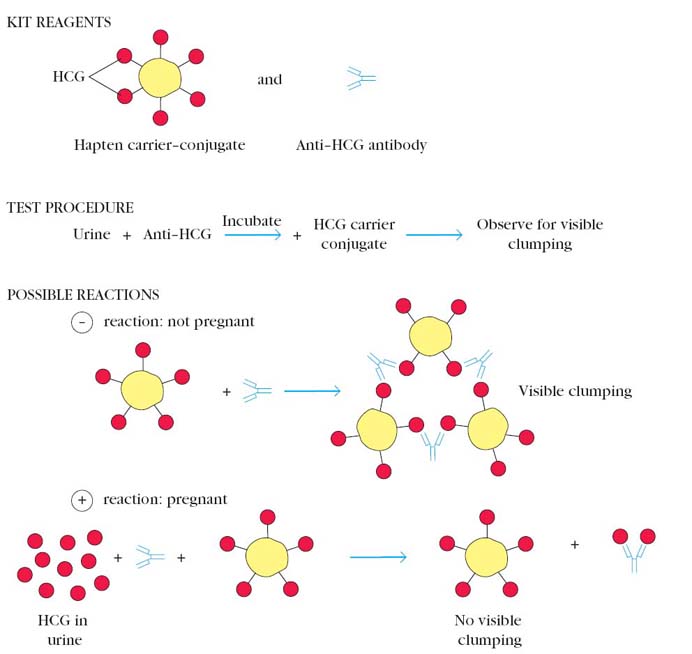
A modification of the agglutination reaction, called agglutination inhibition, provides a highly sensitive assay for small quantities of an antigen. For example, one of the early types of home pregnancy test kits included latex particles coated with human chorionic gonadotropin (HCG) and antibody to HCG. The addition of urine from a pregnant woman, which contained HCG, inhibited agglutination of the latex particles when the anti-HCG antibody was added; thus the absence of agglutination indicated pregnancy.
Agglutination inhibition assays can also be used to determine whether an individual is using certain types of illegal drugs, such as cocaine or heroin. A urine or blood sample is first incubated with antibody specific for the suspected drug. Then red blood cells (or other particles) coated with the drug are added. If the red blood cells are not agglutinated by the antibody, it indicates the sample contained an antigen recognized by the antibody, suggesting that the individual was using the illicit drug. One problem with these tests is that some legal drugs have chemical structures similar to those of illicit drugs, and these legal drugs may cross-react with the antibody, giving a false-positive reaction. For this reason a positive reaction must be confirmed by a nonimmunologic method. Agglutination inhibition assays are widely used in clinical laboratories to determine whether an individual has been exposed to certain types of viruses that cause agglutination of red blood cells. If an individual’s serum contains specific antiviral antibodies, then the antibodies will bind to the virus and interfere with hemagglutination by the virus. This technique is commonly used in premarital testing to determine the immune status of women with respect to rubella virus. The reciprocal of the last serum dilution to show inhibition of rubella hemagglutination is the titer of the serum. A titer greater than 10 (1:10 dilution) indicates that a woman is immune to rubella, whereas a titer of less than 10 is indicative of a lack of immunity and the need for immunization with the rubella vaccine.
ANTIGLOBULIN TEST (COOMBS TEST):
The antiglobulin test was devised by Coombs, Mourant and Race for the detection of anti-Rh antibodies that do not agglutinate Rh positive erythrocytes in saline. When sera containing incomplete anti-Rh antibodies are mixed with Rh positive red cells, the antibody globulin coats the surface of the erythrocytes, though they are not agglutinated. When such erythrocytes coated with the antibody globulin are washed free of all unattached protein and treated with a rabbit antiserum against human gammaglobulin (antiglobulin or Coombs serum), the cells are agglutinated. This is the principle of the antiglobulin test.
The Coombs test may be of the direct or the indirect type. In the direct Coombs test, the sensitization of the erythrocytes with incomplete antibodies takes place in vitro, as in the hemolytic disease of the newborn due to Rh incompatibility. When the red cells of erythroblastotic infants are washed free of unattached protein and then mixed with a drop of Coombs serum, agglutination results. For unknown reasons, the direct Coombs test is often negative in hemolytic disease due to ABO incompatibility.
In the indirect Coombs test, sensitization of red cells with the antibody globulin is performed in vitro. Originally employed for detection of anti-Rh antibodies, the Coombs test is useful for the demonstrating any type of incomplete or non-agglutinating antibody as for example, in brucellosis.
2. SECONDARY IMMUNOTECHNIQUES:
In these techniques, antigen-antibody complexes are identified by using labels like enzyme, radioisotopes, fluorescent substances etc.
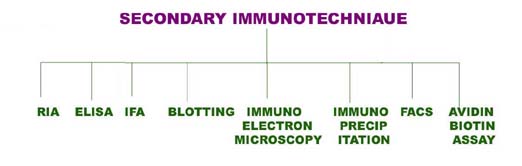
i) Radio Immuno Assay (RIA):
One of the most sensitive technique for detecting antigen or antibody is RIA. This technique was first developed by S.A. Berson and Rosalyn Yalow in 1960. The principle of RIA involves competitive binding of radiolabeled antigen and unlabeled antigen to a high affinity antibody. The labeled antigen is mixed with antibody at a concentration that saturates the antigen binding sites of the antibody molecule and then increasing amount of test sample containing unlabeled antigen of unknown concentration are added. The decrease in amount of radiolabeled antigen bound to specific antibody in the presence of test sample is measured in order to determine the amount of antigen. There are two types of RIA namely, Soluble RIA and Solid-phase RIA.
Soluble RIA:
The antigen is generally labeled with a gamma-emitting isotope such as 125I, but beta-emitting isotopes such as tritium (3H) are also routinely used as labels. The radiolabeled antigen is part of the assay mixture; the test sample may be a complex mixture, such as serum or other body fluids, that contains the unlabeled antigen. The first step in setting up an RIA is to determine the amount of antibody needed to bind 50%–70% of a fixed quantity of radioactive antigen (Ag*) in the assay mixture. This ratio of antibody to Ag* is chosen to ensure that the number of epitopes presented by the labeled antigen always exceeds the total number of antibody binding sites. Consequently, unlabeled antigen added to the sample mixture will compete with radiolabeled antigen for the limited supply of antibody. Even a small amount of unlabeled antigen added to the assay mixture of labeled antigen and antibody will cause a decrease in the amount of radioactive antigen bound, and this decrease will be proportional to the amount of unlabeled antigen added. To determine the amount of labeled antigen bound, the Ag-Ab complex is precipitated to separate it from free antigen (antigen not bound to Ab), and the radioactivity in the precipitate is measured. A standard curve can be generated using unlabeled antigen samples of known concentration (in place of the test sample), and from this plot the amount of antigen in the test mixture may be precisely determined.
Several methods have been developed for separating the bound antigen from the free antigen in RIA. One method involves precipitating the Ag-Ab complex with a secondary anti-isotype antiserum. For example, if the Ag-Ab complex contains rabbit IgG antibody, then goat anti-rabbit IgG will bind to the rabbit IgG and precipitate the complex. Another method makes use of the fact that protein A of Staphylococcus aureus has high affinity for IgG. If the Ag-Ab complex contains an IgG antibody, the complex can be precipitated by mixing with formalin-killed S. aureus. After removal of the complex by either of these methods, the amount of free labeled antigen remaining in the supernatant can be measured in a radiation counter; subtracting this value from the total amount of labeled antigen added yields the amount of labeled antigen bound.
Solid-phase RIA:
Various solid-phase RIAs have been developed that make it easier to separate the Ag-Ab complex from the unbound antigen. In some cases, the antibody is covalently crosslinked to Sepharose beads. The amount of radiolabeled antigen bound to the beads can be measured after the beads have been centrifuged and washed. Alternatively, the antibody can be immobilized on polystyrene or polyvinylchloride wells and the amount of free labeled antigen in the supernatant can be determined in a radiation counter.
In another approach, the antibody is immobilized on the walls of microtiter wells and the amount of bound antigen determined. Because the procedure requires only small amounts of sample and can be conducted in small 96-well microtiter plates (slightly larger than a 3 _ 5 card), this procedure is well suited for determining the concentration of a particular antigen in large numbers of samples. For example, a microtiter RIA has been widely used to screen for the presence of the hepatitis B virus. RIA screening of donor blood has sharply reduced the incidence of hepatitis B infections in recipients of blood transfusions.
ii) Enzyme immuno assay (EIA):
Enzymes labeled conjugates were introduced first in 1966 for localization of antigens in tissues, as an alternative for fluorescent conjugates. In 1971, enzyme labeled antigens and antibodies were developed as serological reagents for assay of antibodies and antigens. Their versatility, sensitivity, simplicity, economy and absence of radiohazard have made EIA the most widely used procedure in clinical serology. The availability of test kits ad facility for automation has added to their popularity.
The term enzyme immunoassay (EIA) includes all assays based on the measurement of enzyme labeled antigen, hapten or antibody. EIAs are of two basic types namely homogenous and heterogeneous. In homogenous EIA, there is no need to separate the bound and free fractions so that the test can be completed in one step, with all reagents added simultaneously. This type of EIA can be used only for assay of haptens such as drugs and not for microbial antigens and antibodies. An example of homogenous EIA is Enzyme multiplied immunoassay technique (EMIT), which is a simple assay method for small molecule drug such as opiates, cocaine, barbiturates or amphetamine in serum. In this technique, enzyme become active only when it binds to its target molecule and that is why there is no need to separate enzyme and reaction mixture.
Heterogeneous EIA requires the separation of the free and bound fraction either by centrifugation or by adsorption on solid surfaces and washing. It is therefore a multistep procedure, with reagents added sequentially. The major type of heterogenous EIA is enzyme linked immunosorbent assay (ELISA).
Enzyme Linked Immunosorbent Assay (ELISA):
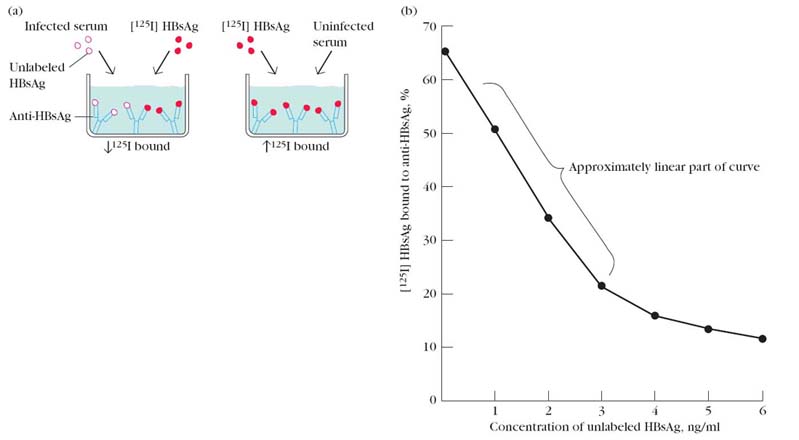
Enzyme-linked immunosorbent assay, commonly known as ELISA (or EIA), is similar in principle to RIA but depends on an enzyme rather than a radioactive label. An enzyme conjugated with an antibody reacts with a colorless substrate to generate a colored reaction product. Such a substrate is called a chromogenic substrate. A number of enzymes have been employed for ELISA, including alkaline phosphatase, horseradish peroxidase, and b-galactosidase. These assays approach the sensitivity of RIAs and have the advantage of being safer and less costly. There are three types of ELISA, namely Indirect, Sandwitch and Competitive ELISA.
INDIRECT ELISA
Antibody can be detected or quantitatively determined with an indirect ELISA. Serum or some other sample containing primary antibody (Ab1) is added to an antigen-coated microtiter well and allowed to react with the antigen attached to the well. After any free Ab1 is washed away, the presence of antibody bound to the antigen is detected by adding an enzyme-conjugated secondary anti-isotype antibody (Ab2), which binds to the primary antibody. Any free Ab2 then is washed away, and a substrate for the enzyme is added. The amount of colored reaction product that forms is measured by specialized spectrophotometric plate readers, which can measure the absorbance of all of the wells of a 96-well plate in seconds. Indirect ELISA is the method of choice to detect the presence of serum antibodies against human immunodeficiency virus (HIV), the causative agent of AIDS. In this assay, recombinant envelope and core proteins of HIV are adsorbed solid-phase antigens to microtiter wells. Individuals infected with HIV will produce serum antibodies to epitopes on these viral proteins. Generally, serum antibodies to HIV can be detected by indirect ELISA within 6 weeks of infection.
SANDWICH ELISA
Antigen can be detected or measured by a sandwich ELISA. In this technique, the antibody (rather than the antigen) is immobilized on a microtiter well. A sample containing antigen is added and allowed to react with the immobilized antibody. After the well is washed; a second enzyme-linked antibody specific for a different epitope on the antigen is added and allowed to react with the bound antigen. After any free second antibody is removed by washing, substrate is added, and the colored reaction product is measured.
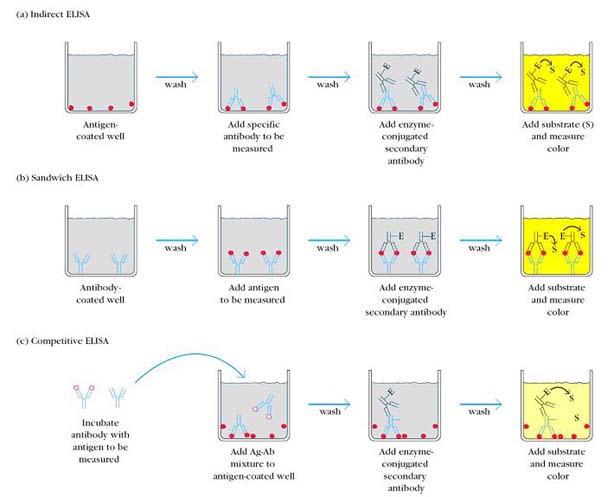
COMPETITIVE ELISA
Another variation for measuring amounts of antigen is competitive ELISA. In this technique, antibody is first incubated in solution with a sample containing antigen. The antigen-antibody mixture is then added to an antigen coated microtiter well. The more antigens present in the sample, the less free antibody will be available to bind to the antigen-coated well. Addition of an enzyme-conjugated secondary antibody (Ab2) specific for the isotype of the primary antibody can be used to determine the amount of primary antibody bound to the well as in an indirect ELISA. In the competitive assay, however, the higher the concentration of antigen in the original sample, the lower the absorbance.
CASSETTE ELISA:
A simple modification of ELISA which has found wide application for testing one or a few samples of sera at a time is the cylinder or cassette ELISA. Here each specimen is tested in a separate disposable cassette. The test is rapid, taking only about 10 minutes as compared to 2-4hours which are taken for microplate ELISA. There is no need for microplate washers or readers. The result is read visually. Inbuilt positive and negative controls are usually provided for validation of the test procedure.
An example of cassette ELISA is the one used for the detection of HIV type 1 and 2 antibodies. Specific type 1 and 2 antigens are immobilized at separate fixed sites on the nitrocellulose membrane in the cassette. Test serum is added on the membrane and allowed to filter into absorbent material placed below it in the cassette base. Antibody, if present in the serum will bind to the appropriate antigen. After washing to remove unbound antibody, enzyme labeled antihuman immunoglobulin antibody is added. After additional washing to remove unbound conjugate, a substrate yielding a colored product is added. A positive result is indicated by a colored spot developing at the site of the antigen against which antibody is present in the serum. Human immunoglobulin immobilized at a spot on the membrane acts as a control for the test procedure, as shown by the development of the color at the site.
iii) Immunofluorescence:
Fluorescence is the property of absorbing light rays of one particular wavelength and emitting rays with a different wavelength. Fluorescent dyes show up brightly under ultraviolet light as they convert ultraviolet into visible light. Coons and his colleagues (1942) showed that fluorescent dyes can be conjugated to antibodies and that such labeled antibodies can be used to locate and identify antigens in tissue. Because of this reason, immunofluorescence technique is also a type of immunohistochemistry assay.
The most commonly used fluorescent dyes are fluorescein and rhodamine but other highly fluorescent substances such as phycoerythrin and phycobiliproteins have also been used. Dyes can be conjugated to the Fc region of an antibody molecule without affecting the specificity of antibody. Fluorescein, an organic dye that is the most widely used label for immunofluorescence procedures absorb blue light (490nm) and emits an intense yellow green fluorescence (517nm). Rhodamine, another organic dye absorb in the yellow green range (515nm) and emits a deep red fluorescence (546nm).
Fluorescent-antibody staining of cell membrane molecules or tissue sections can be direct or indirect. In direct staining, the specific antibody (the primary antibody) is directly conjugated with fluorescein; in indirect staining, the primary antibody is unlabeled and is detected with an additional fluorochrome-labeled reagent. A number of reagents have been developed for indirect staining. The most common is a fluorochrome-Labeled secondary antibody raised in one species against antibodies of another species, such as fluorescein-labeled goat anti-mouse immunoglobulin.
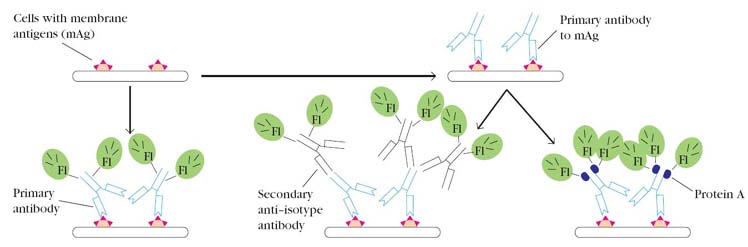
Indirect immunofluorescence staining has two advantages over direct staining. First, the primary antibody does not need to be conjugated with a fluorochrome. Because the supply of primary antibody is often a limiting factor, indirect methods avoid the loss of antibody that usually occurs during the conjugation reaction. Second, indirect methods increase the sensitivity of staining because multiple molecules of the fluorochrome reagent bind to each primary antibody molecule, increasing the amount of light emitted at the location of each primary antibody molecule. In indirect staining procedure, Protein A molecule labeled with flurochrome used to detect the primary antibodies. The protein A is obtained from cell wall of staphylococcus aureus. Its ability to bind with Fc region of immunoglobulins utilized in this assay procedure. It might also used in other techniques like RIA and ELISA instead of secondary antibodies. Immunofluorescence has been applied to identify a number of subpopulations of lymphocytes, notably the CD4 and CD8 T-cell subpopulations. The technique is also suitable for identifying bacterial species, detecting Ag-Ab complexes in autoimmune disease, detecting complement components in tissues, and localizing hormones and other cellular products stained in situ. Indeed, a major application of the fluorescent-antibody technique is the localization of antigens in tissue sections or in subcellular compartments. Because it can be used to map the actual location of target antigens, fluorescence microscopy is a powerful tool for relating the molecular architecture of tissues and organs to their overall gross anatomy.
iv). Western Blotting:
Identification of a specific protein in a complex mixture of proteins can be accomplished by a technique known as Western blotting, named for its similarity to Southern blotting, which detects DNA fragments, and Northern blotting, which detects mRNAs.
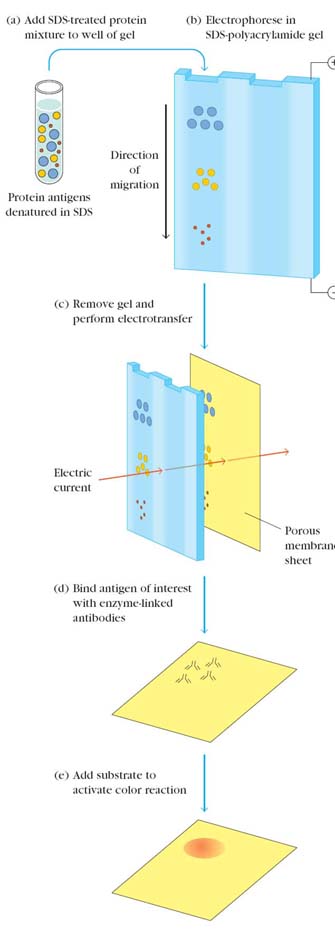
In Western blotting, a protein mixture is electrophoretically separated on an SDS-polyacrylamide gel (SDS-PAGE), a slab gel infused with sodium dodecyl sulfate (SDS), a dissociating agent. The protein bands are transferred to a nylon membrane by electrophoresis and the individual protein bands are identified by flooding the nitrocellulose membrane with radiolabeled or enzymelinked polyclonal or monoclonal antibody specific for the protein of interest. The Ag-Ab complexes that form on the band containing the protein recognized by the antibody can be visualized in a variety of ways. If the protein of interest was bound by a radioactive antibody, its position on the blot can be determined by exposing the membrane to a sheet of x-ray film, a procedure called autoradiography. However, the most generally used detection procedures employ enzyme-linked antibodies against the protein. After binding of the enzymeantibody conjugate, addition of a chromogenic substrate that produces a highly colored and insoluble product causes the appearance of a colored band at the site of the target antigen. The site of the protein of interest can be determined with much higher sensitivity if a chemiluminescent compound along with suitable enhancing agents is used to produce light at the antigen site.
Western blotting can also identify a specific antibody in a mixture. In this case, known antigens of well-defined molecular weight are separated by SDS-PAGE and blotted onto nitrocellulose. The separated bands of known antigens are then probed with the sample suspected of containing antibody specific for one or more of these antigens. Reaction of an antibody with a band is detected by using either radiolabeled or enzyme-linked secondary antibody that is specific for the species of the antibodies in the test sample. The most widely used application of this procedure is in confirmatory testing for HIV, where Western blotting is used to determine whether the patient has antibodies that react with one or more viral proteins.
v). Immunoelectron Microscopy:
The fine specificity of antibodies has made them powerful tools for visualizing specific intracellular tissue components by immunoelectron microscopy. In this technique, an electron-dense label is either conjugated to the Fc portion of a specific antibody for direct staining or conjugated to an antiimmunoglobulin reagent for indirect staining. A number of electron-dense labels have been employed, including ferritin and colloidal gold. Because the electron-dense label absorbs electrons, it can be visualized with the electron microscope as small black dots. In the case of immunogold labeling, different antibodies can be conjugated with gold particles of different sizes, allowing identification of several antigens within a cell by the different sizes of the electron-dense gold particles attached to the antibodies.
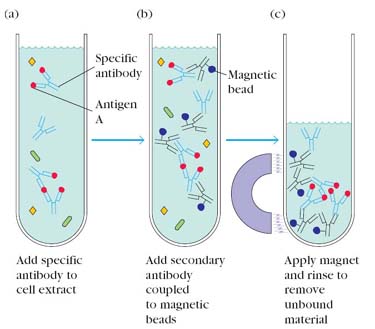
vi). Immunoprecipitation:
The immunoprecipitation technique has the advantage of allowing the isolation of the antigen of interest for further analysis. It also provides a sensitive assay for the presence of a particular antigen in a given cell or tissue type. In this technique, test sample taken in test tube and add antibody to that of specific antigen to be isolated or determined. The antigen-antibody complex is formed and then add secondary antibody which contain magnetic beads against primary antibody which already bound to antigen. After secondary antibody binds to primary antibody, immunoprecipitation achieved by passing a magnet against the sides of the test tube. Immunoprecipitates then utilized for further studies. Immunoprecipitatin is used to determine whether a particular antigen is actually synthesized by a cell or tissue.
vii) Flowcytometry:
This technique otherwise known as Fluorescence Activated Cell Sorting (FACS). The fluorescent antibody techniques described are extremely valuable qualitative tools, but they do not give quantitative data. This shortcoming was remedied by development of the flow cytometer, which was designed to automate the analysis and separation of cells stained with fluorescent antibody. This FACS uses a laser beam and light detector to count single intact cells in suspension.
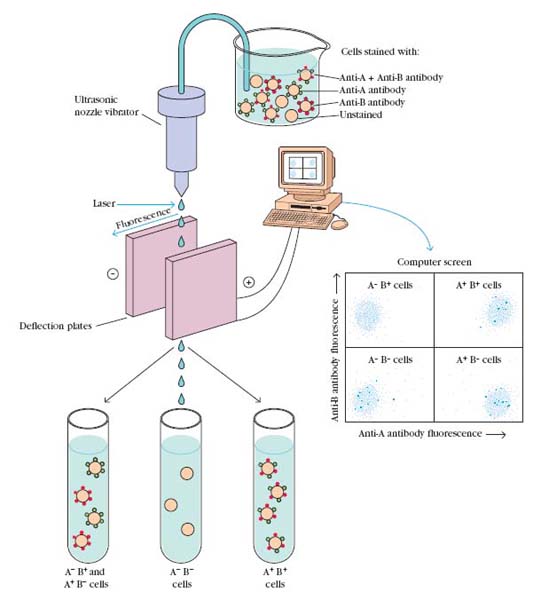
In this technique, a mixed cell population is stained with two antibodies, one specific for surface Ag-A and other specific for Ag-B. The antibody against Ag-A labeled with fluorescein and antibody against Ag-B labeled with rhodamine. The labeled cells are mixed with sheath fluid and placed in vibrating chamber. The vibration of chamber adjusted such a manner so that the cells are expelled one at a time from a small vibrating nozzle that generates micro droplets each containing not more than a single cell. The flow cytometer uses a laser beam and light detector to count single intact cells in suspension. Every time a cell passes the laser beam, light is deflected from the detector, and this interruption of the laser signal is recorded. Those cells having a fluorescently tagged antibody bound to their cell surface antigens are excited by the laser and emit light that is recorded by a second detector system located at a right angle to the laser beam. The intensity of the fluorescence emitted by each droplet that contains a cell is monitered by a detector and displayed on a computer screen. Because the computer tracks the position of each droplet, it is possible to determine when a particular droplet will arrive between the deflection plates. By applying a momentary charge to the deflection plates when a droplet is passing between them, it is possible to deflect the path of a particular droplet into one or another collecting vessel. This allows the sorting of a population of cells into subpopulations having different profiles of surface markers. In the computer displays each dot represents a cell.
The FACS has multiple applications. It is most commonly used to determine the kind and number of white blood cells in each population in patient’s blood sample. This technique is also used for the ell counting in a mixture of cells. FACS also used to find out the cell shape and cell size.
Flow cytometry now occupies a key position in immunology and cell biology, and it has become an indispensable clinical tool as well. In many medical centers, the flow cytometer is one of the essential tools for the detection and classification of leukemias. The choice of treatment for leukemia depends heavily on the cell types involved, making precise identification of the neoplastic cells an essential part of clinical practice. Likewise, the rapid measurement of T-cell subpopulations, an important prognostic indicator in AIDS, is routinely done by flowcytometric analysis. In this procedure, labeled monoclonal antibodies against the major T-cell subtypes bearing the CD4 and CD8 antigens are used to determine their ratios in the patient’s blood. When the number of CD4 T cells falls below a certain level, the patient is at high risk for opportunistic infections.
viii) Avidin-Biotin Assay:
In some instances direct labeling of proteins, especially with enzymes or other large molecules, may cause denaturation and loss of activity. A convenient labeling system has been developed which may be used in conjunction with the ELISA and ELISPOT assays. This labeling technique exploits the high affinity of the reaction between the vitamin biotin and avidin, a large molecule that may be labeled with radioactive isotopes, with fluorescent molecules, or with enzymes. Biotin is a small molecule (mol.wt. 244) that can be coupled to an antibody (or to any protein molecule) by a gentle chemical reaction that causes no loss of antibody activity. After the biotin-coupled antibody has reacted in the assay system, the labeled avidin is introduced and binding is measured by detecting the label on the avidin molecule. The reaction between biotin and avidin is highly specific and of such high affinity that the bond between the two molecules under most assay conditions is virtually irreversible.
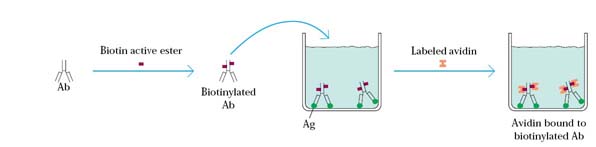
The source for the avidin was egg white. But recently it was found that even microorganism like streptomyces avidinii, produces streptavidin which act like avidin. Because of this, avidin available easily.