Clinical Drug Development
Introduction
After the discovery of a drug candidate, there remain a number of
hurdles to overcome before it can be marketed. The ultimate goal is to prove
that the drug is safe and efficacious. The watershed between research and
development has been reached. Here the overall process of evaluation of the drug
candidate changes. The chemistry group and other scientists provide the
development group with an opportunity to take to the clinic a chemical with
therapeutic utility. The mandate of the development team is to take the chemical
entity and change it into a drug that regulatory authorities are prepared to
license. In any pharmaceutical company, big or small, transitioning from
research to development is probably the largest paradigm shift. The laws of
science are supplemented with three additional primarily nonscientific
disciplines, adherence to the ethical code of medical research, proof of
efficacy and safety, and compliance with regulatory requirements.
Scientific discipline and development discipline are horses of a
different color. In research it is a requirement to get the “best possible”
entity; in development it is the requirement to choose the potential drug that
will be proved to have therapeutic utility in a reasonable development time
frame and will be approved by regulatory authorities to be marketed. Research is
always looking for the “best”; development is always looking for the “best
registrable.” The rules governing the two disciplines are separated by a
bureaucratic chasm that requires the research group to be acceptant of a
significant up-regulation of bureaucratic process. Development requires much
more red tape.
History of Ethical Medical Research
Drug development is wrapped in two interlocking but independent
disciplines. On the one hand, there are the regulatory and quasiscientific
disciplines, which are dealt with later in this chapter, and on the other hand,
there is the moral responsibility for the subject, the ethics of clinical
research. Clinical research was being conducted long before there were moves to
require proof of safety and efficacy before a drug could be put on the market.
In the early days of medical research, ethics were considered to be covered by
the medical treatment ethical codes and were not considered as a separate
entity. Possibly the best known early medical ethics code was from ancient
Greece, the Hippocratic Oath, a modernized version of which is still in use. The
American Medical Association’s current
Code of Medical Ethics
states that the Hippocratic Oath
“is an expression of ideal conduct for the physician.” The oath in its original
version referred exclusively to the role of the physician in patient management
and therefore is not directly relevant to research. However, the oath contained
the statement, “I shall keep them from harm and injustice,” a sentiment that
encapsulates the essence of the more modern codes of ethics.
There is some controversy over who was the original pioneer of
clinical research ethics. One school cites Thomas Percival, an Edinburgh
University, Scotland–trained English physician (1740–1804), as the originator of
clinical research ethics. He published an ethical code of conduct in 1794,
Medical Ethics or a Code of Institutes and Precepts
Adapted to the Professional Conduct of Physicians and Surgeons.
This code was adapted by the American Medical
Association for use by American physicians in 1847. Percival’s detractors point
out that the Percivilian code focused on the responsibility of physicians to
care for the sick. Although it contained guidance on the use of experimental
techniques on patients (“[these] should be scrupulously and conscientiously
governed by sound reason”), it did not mention one of the essential modern-day
concepts, the consent of the subject. Another school cites William Beaumont, a
Connecticut-born physician (1785–1853) and sometime army surgeon who became
known as the “father of gastric physiology.” It is said that in 1833 he espoused
an ethical research code which included the requirement for voluntary consent by
any research subject, cessation of the research if it caused distress to the
subject, and the provision that the subject was free to withdraw from the
research whenever he or she wanted. Beaumont’s detractors question whether this
code ever existed. Whoever has the greater claim to be the father of clinical
research ethics, it is apparent that the basic precepts of clinical research
ethics were laid down over 100 years before the barbaric deviation from ethical
conduct which led to the creation of today’s clinical research ethics.
The modern era in medical research ethics began with the
Nuremberg Code (1947). It is no small irony that the clearest pronouncements on
the pivotal role of consent were promulgated in Germany at the beginning of the
twentieth century. Indeed, in 1931 the German Reichs Minister of the Interior
forbad medical experimentation unless the “subject or his legal representative
has unambiguously consented to the procedure in the light of relevant
information provided in advance.” Just 15 years later, over 20 physicians who
worked for the German armed forces stood trial for atrocities committed during
World War II. Some were sentenced to death, most because they had conducted
human medical experimentation to which no sane person would have consented.
Germany did not hold the monopoly of unethical research. The Office of
Scientific Research and Development in the United States was accused of using
subjects for experimentation without them giving informed consent.
The Nuremberg Code of 1947 defined the voluntary consent of the
subject in the following terms; This means that the person involved should have
legal capacity to give consent; should be so situated as to be able to exercise
free power of choice … and should have suffi cient knowledge and comprehension
of the elements of the subject matter involved as to enable him to make an
understanding and enlightened decision. This latter element requires that before
the acceptance of an affirmative decision by the experimental subject there
should be known to him the nature, duration and purpose of the experiment; the
methods and means by which it is to be conducted; all inconveniences and hazards
reasonably to be expected; and the effects upon his health or person which may
possibly come from his participation in the experiment. This clear definition of
informed consent was supplemented by the following principles:
• Investigators must be scientifically qualified.
• Research must be purposeful and necessary for the benefit of
society.
• Appropriate measures should be taken to avoid or protect
subjects from injury or unnecessary
physical or mental suffering.
• The risks to the subjects shall not be greater than the
humanitarian importance of the
problem.
• Subjects may terminate the experiment at any time.
• Research must be based on animal studies or other rational
justification.
The Nuremberg Code encompasses all that has to be adhered to for
appropriate clinical research to be conducted without harming the subject who
volunteers. The field of clinical research ethics is no more immune from the
reinvention of the wheel than any other quasiscientific discipline. The desire
for improvement is both laudable and counterproductive. It is fueled by the
reality of policy versus practice.
Porton Down in the UK was a biochemical research facility. In the
mid-1950s, part of its research activities were concerned with the effects of
nerve gases. One young serviceman was asked to participate in an experiment to
find a cure for the common cold. It is claimed that he was actually exposed to a
nerve toxin, sarin, and subsequently died. As recently as 2002 the British High
Court gave the go-ahead for a new inquest into the death. Ethical issues in
medical research have a habit of not going away until there is complete
resolution.
Some experiments that were on shaky or clearly nonethical grounds
at their inception lived on even after the adoption of the Nuremberg Code.
Probably the best known is the Tuskegee Syphilis Study. In 1932, three hundred
and ninety-nine African Americans who were diagnosed as suffering from syphilis
were entered into the study, as were 200 healthy African Americans who acted as
controls. There was no informed consent. The purpose of the study was to prove
that the antisyphilitic medications available at the time were not just
ineffective but were harmful. The requirement to generate data to prove this
hypothesis might be considered laudable, but the methods used to populate the
study were not ethical. After the study started, the way that the participants
were managed raised major ethical concerns. By 1947, penicillin was considered a
safe and effective treatment for syphilis. The Tuskegee syphilis patients were
not evaluated as to whether they might benefit from this new medication. One
justification was that their disease was too advanced for them to be candidates
for benefit. The study continued and would no doubt have ended only when all 599
participants had died had its existence not been leaked to the press in 1970. In
1972, an advisory panel determined that the study was medically unjustified. The
study was closed and a class action law suit led to a multimillion dollar
restitution to the survivors and the surviving family members of this misguided
and unethical study. In 1997, President Clinton formally apologized to the
Tuskegee study participants.
With human rights abuses such as these, there is more than enough
justification to continue to legislate against abuse or at least to try to defi
ne what is unethical. The Nuremberg Code formed the basis of the Declaration of
Geneva Physician’s Oath (1948). This was adopted by General Assembly of the
newly formed World Medical Association (WMA). It was looked on as a
modernization of the Hippocratic Oath and was an attempt to focus the individual
physician’s attention on medical ethics.
Physician’s oath
At the time of being admitted as a member of the medical
profession:
• I solemnly pledge myself to consecrate my life to the service
of humanity.
• I will give to my teachers the respect and gratitude which is
their due.
• I will practice my profession with conscience and dignity; the
health of my patient will be
my first consideration.
• I will maintain by all the means in my power, the honor and
noble traditions of the medical
profession; my colleagues will be my brothers.
• I will not permit considerations of religion, nationality,
race, party politics or social standing
to intervene between my duty and my patient.
• I will maintain the utmost respect for human life from the time
of conception, even under
threat, I will not use my medical knowledge contrary to the laws
of humanity.
• I make these promises solemnly, freely and upon my honor.
The WMA went on to adopt an International Code of Medical Ethics
(1949). This was an attempt to develop international standards of medical ethics
and sought to summarize the most important principles. It did not specifically
address clinical research ethics. That topic was brought to the attention of the
WMA Medical Ethics Committee in 1953. After several years of discussion and
research a draft declaration was finally tabled in 1961. It was adopted at the
18th WMA General Assembly, held in Helsinki in 1964. The main points in the
declaration are:
• Clinical research should be based on adequately performed
laboratory and animal
experimentation.
• It must be conducted by scientifically qualified persons.
• There must be a protocol which will be reviewed by an ethical
committee.
• The risk-benefit ratio must be favorable.
• Informed consent must be obtained.
The Declaration of Helsinki was the first time that medical
ethics impinged on the regulation of new drugs. The first item could be said to
be the start of the requirement for documentation to establish that sufficient
preclinical research has been conducted to allow a drug to be given to a human
being. In the United States, this documentation is called the investigational
new drug (IND) application. It could be argued that its existence is due largely
to the ethical considerations that were behind the Declaration of Helsinki. From
this requirement there grew a massive industry dedicated to preclinical research
in animals. This has led to additional ethical considerations regarding the use
of animals in the development of a drug. At one extreme it is argued that the
use of animals in biomedical research is unnecessary because equivalent
information can be obtained by alternative methods. The fact is that it may be
possible sometime in the future, but is not at present. Some animal studies that
were at one time considered essential for an IND, such as the LD50 (the lethal
dose of drug required to kill 50% of the animals to which it is given), have
been reevaluated and dropped from the preclinical requirements. Nonanimal
evaluation can supplement animal testing and can reduce the use of animals in
research (e.g., in the UK the number of laboratory animals used annually has
almost halved in the past 20 years), but the technology does not exist to
supplant animal studies completely. Relevant research requires intact
physiological systems, and they cannot be mimicked accurately at present. It is
not acceptable ethically to risk the health of a human being on the basis of
nonphysiological data alone. The Declaration of Helsinki remained unchanged
until 1975 when an extensive revision, conducted on behalf of the WMA, was
adopted at the 29th General Assembly in Tokyo. This amendment included an
expansion of the basic principles and categories developed to address clinical
research combined with therapeutic care and clinical research for purely
scientific purposes.
Further revisions occurred in 1983 (Venice amendment), 1989 (Hong
Kong amendment), and 1996 (Somerset West, Republic of South Africa, amendment).
The last of these revisions caused something of an uproar in the medical
research community. What made the balloon go up was the language of paragraph
29: “The benefits, risks, burdens and effectiveness of a new method should be
tested against those of the best current prophylactic, diagnostic and
therapeutic methods.
This does not exclude placebo,
or no treatment, in studies where no proven prophylactic, diagnostic or
therapeutic method exists”
[author’s italics]. To put it another way, a placebo-controlled study design
could not be used ethically in drug studies unless no other treatment was
available. This interpretation effectively excluded the placebo-controlled study
from clinical research. Most regulatory authorities considered that the
placebo-controlled clinical trial design was vital to the evaluation of drug
efficacy and safety in the majority of medical conditions. Here again, as with
the advent of the Declaration of Helsinki, medical ethics impinged on clinical
research and drug regulatory process. It took four years before that controversy
was partially resolved. In 2000 at the General Assembly in Edinburgh, Scotland a
note of clarification was attached to the Declaration of Helsinki;
it read:
[A] placebo controlled trial may be ethically acceptable, even if
proven therapy is available, under the following circumstances:
• Where for compelling and scientifically sound methodological
reasons its use is necessary to determine the efficacy or safety….
• Where a prophylactic, diagnostic or therapeutic method is being
investigated for a minor condition and the patients who receive placebo will not
be subject to any additional risk of serious or irreversible harm.
This masterpiece of bureaucratic language was meant to blunt the
debate, but the controversy
still has not gone away. The Declaration of Helsinki continues to
evolve and its presence will ensure that medical ethics and clinical research
will forever be intertwined. In the United States it was as a direct result of
the revelation of the Tuskegee Syphilis Study that the next U.S. medical ethics
initiative emerged. The National Research Act of 1974 was passed (Public Law
93348), which required regulatory protection for human subjects and created the
National Commission for the Protection of Human Subjects of Biomedical and
Behavioral Research. In 1979 this commission produced the Belmont Report, named
after the Smithsonian Institution’s Conference Center, where the discussions
were first held in 1976. The report established three ethical principles to
allow problems to be solved in the area of ethics in clinical research:
(1) respect for persons, (2) beneficence, and (3) justice.
In general terms, these categories were equivalent to informed
consent, risk–benefit assessment, and an appropriate choice of subjects for the
research. U.S. federal regulations were developed from the Belmont Report. They
were adopted, in 1991 by 17 federal departments and agencies: hence the term
“the Common Rule.” This governs research conducted or supported by these
departments and agencies. The regulations are called Title 45, Code of Federal
Regulations 46 (45 CFR 46): Federal Policy for the Protection of Human Subjects.
There are four parts:
1.
Subpart A: Department of Health and Human Services (DHHS)
policy for the protection of human research subjects
2.
Subpart B: DHHS protections pertaining to research,
development, and related activities involving fetuses, pregnant women, and human
in vitro fertilization
3.
Subpart C: DHHS protections pertaining to biomedical and
behavioral research involving prisoners as subjects
4.
Subpart D: DHHS protections for children involved as subjects
in research
The Food and Drug Administration (FDA) has a different set of
regulations governing human research, including:
• 21 CFR 50: Informed Consent
• 21 CFR 56: Institutional Review Boards
• 21 CFR 312: Investigational New Drug Application
All clinical research is subject to these and other FDA
regulations. Medical ethical issues are now enshrined in U.S. law. Let the final
word on ethics come from the National Bioethics Advisory Commission: “It is
essential that the research community come to value the ethics of research as
central to the scientific process.”
History of the Regulation of Medical Research
Today, there is general acceptance that clinical testing of
proposed therapeutic entities is mandatory. No drug will be approved without
compelling clinical evidence of safety and efficacy. Clinical studies cannot be
initiated without a clearly defi ned preclinical development
program which forms an integral part of the IND.
At the beginning of the twentieth century, preclinical testing of
drugs was not obligatory, and the requirement of clinical trials to demonstrate
that a drug was safe, let alone efficacious, had never been seriously
considered. In the final years of the nineteenth century, Dr. Harvey W. Wiley
was appointed chief chemist at the Department of Agriculture.
He embarked upon a crusade to protect the public from adulterated
food and established in 1903 a volunteer “poison squad,” who agreed to eat food
that was treated with chemical preservatives to establish whether they were
injurious to health. Among the chemicals fed to the poison squad were salicylic
acid, formaldehyde, benzoic acid, and borax. After five years it was concluded
that chemical preservatives should be used in foods only when necessary, a
sentiment with which few would argue. Wiley expanded his interest to drugs, and
by persistent lobbying and campaigning was a major force behind the inclusion of
provisions to protect the public against “misbranded” or adulterated drugs in
the Pure Food and Drug Act signed by President Theodore Roosevelt in 1906. This
act was concerned only with violations of the food and drug regulations after
they occurred. There were no provisions for testing new drugs. Three decades
later the act was still in force. There were no requirements to determine the
clinical safety of drugs. It took a major disaster in health care to set in
motion the regulatory processes that we consider indispensable today to ensure
patients’ safety.
In 1932, Gerhard Domagk demonstrated that a chemical called
prontesil protected mice against some bacterial infections. Subsequent
evaluation showed that prontesil was metabolized to
p-aminobenzenesulfonamide,
which was known as sulfanilamide. As prontesil had been discovered in 1908,
there were no intellectual property issues. Many pharmaceutical companies,
including Merck, Parke-Davis, and Eli Lilly, had obtained the backing of the
American Medical Association Council on Pharmacy and Chemistry (AMACPC) to
market sulfanilamide in capsules and tablets for streptococcal infections. It
should be noted that this AMACPC review was not a legal requirement before a
drug could be marketed. In 1937 sulfanilamide was being used extensively in the
treatment of a variety of infectious diseases. A small pharmaceutical company,
S. E. Massengill of Bristol, Tennessee, became aware that there was an unmet
need for a liquid preparation. The company’s head chemist was instructed to
develop such a product. The reason that a liquid formulation was not available
was that a suitable solvent had not been identified. The formulation that the
Massengill Company produced comprised diethylene glycol (better known now for
its use as an industrial solvent), water, and flavorings, including raspberry
extract. The liquid formulation was called an
elixir. This term was reserved
exclusively for formulations that contained ethanol. Elixir Sulfanilamide did
not, a fact that played an important role in minimizing an iatrogenic
catastrophe. The liquid formulation of sulfanilamide was “tested”, but the tests
that the company conducted were based on its marketability and included
appearance and flavor acceptability. No toxicity testing was conducted; none was
required by the Food and Drugs Act of 1906.
In the fall of 1937, Massengill’s Elixir Sulfanilamide was
distributed and was used by approximately 350 patients. Nearly one-third of
those patients died, due primarily to renal failure. The Massengill Company’s
response to this disaster was as forceful as it could be; they sent out over
1000 telegrams requesting the return of Elixir Sulfanilamide from the
distributors. As the extent of the tragedy became apparent, a government
department, the FDA, moved with commendable alacrity to seize the remainder of
the manufactured batch. Without a legal nicety in the act of 1906, the FDA would
have been powerless to prevent the distribution of the remainder of the first
manufactured batch. However, as Elixir Sulfanilamide did not contain ethanol, it
was “misbranded.” A misbranded product could be seized. Such was the fi ne line
drawn by bureaucratic language, which prevented a tragedy from becoming a
medical catastrophe. It was the medical enforcement equivalent of Al Capone
being sent to prison for tax evasion rather than bootlegging, racketeering, and
murder. Had the entire batch of Elixir Sulfanilamide been distributed and
consumed, the death toll would have reached several thousand. In Massengill’s
defense, however, it should be stated that they had contravened no laws other
than the issue of misbranding. This medical tragedy produced one positive
outcome. The U.S. Congress was galvanized into passing the Federal Food, Drug
and Cosmetic Act, which was introduced by Senator Royal S. Copeland and signed
by President Franklin Roosevelt in 1938. It replaced the original drug
legislation from 1906. This act began regulation of the pharmaceutical industry.
Drug manufacturers were henceforth required to provide scientific
proof of safety of new drug products before they were allowed to market them. In
addition, proof of fraud was no longer necessary before action could be taken to
prevent false claims being made for drugs. Hitherto, wild exaggerations were
common. Labeling claims for drugs emblazoned with such names as “Warner’s Safe
Cure for Diabetes” could be stopped only if it could be proved that the
manufacturer of the medication did not believe the claim was justified. The
reversal of this absurd interpretation of freedom of speech was a major step
forward in protecting consumers from unsubstantiated claims but fell well short
of protecting the public from being exposed to nonefficacious “snake oil”
products. Proof of efficacy was the next watershed in the protection of the
public from those pharmaceutical manufacturers who were prepared to take the
money but not deliver the therapeutic goods.
The legislation that was to be enacted to require proof of
efficacy was forced on Congress by a most unusual and tragic set of
circumstances. It was recognized by the legislators that additional controls on
pharmaceutical products were required, although their concept of what type of
controls did not assign efficacy a major role. In 1960, Senator Estes Kefauver
initiated hearings to control unfair marketing practices. The main thrust of
what became Kefauver’s bill dealt with pricing and intellectual property. It
paid lip service to the proof of efficacy. The bill was not popularly received
and would probably never have gained sufficient support to be enacted had it not
been for another horrendous medical tragedy. Chemie Grunenthal was a German
company which manufactured a wide variety of over-the-counter and prescription
drugs that were sold by many different companies. One of the drugs that it
manufactured was called thalidomide. It was a tranquilizer that was recommended
for, among other indications, the treatment of morning sickness in pregnant
women. First marketed in the mid-1950s, by 1962 it was on the market in 46
countries. Thalidomide was marketed in West Germany in 1957, and reports started
to be released concerning potentially drug-related neural toxicity. In addition,
there were reports of congenital malformations in babies born to women who had
taken thalidomide. The predominant malformations were limb deformities,
including shortening or missing arms, with hands extending from the shoulders,
and similar problems with legs. This malformation was not unknown; it had been
reported as early as the eighteenth century and was called
phocomelia
after the Greek word for “seal
limbs.” The drug continued to be marketed despite increasing evidence that it
was toxic, because preclinical testing of the drug in pregnant rats, mice,
hamsters, dogs, and primates had not shown this teratogenic potential.
However, in 1962 the drug was withdrawn voluntarily because of
increasingly negative public opinion.
In the United States in 1960, Richardson-Merrell sought marketing
approval for thalidomide under the brand name Kevadon. It never reached the
market because of the resistance of an FDA medical reviewer, Dr. Francis Kelsey.
It is said that she was influenced by her previous experience with the
antimalarial drug quinidine, which had teratogenic activity. Her misgivings were
based on concerns that peripheral neuritis had been observed in adults. This
mixture of concern about safety and previous experience combined to overrule the
considerable body of preclinical evidence that the drug was safe. Kelsey
exercised the bureaucrats’ power to delay the approval process and thereby
prevented a major medical disaster in the United States. It is believed that
mine babies were born with thalidomide-induced phocomelia in the United States,
whereas in the rest of the world the total is conservatively estimated at
10,000. As a result of her actions, Kelsey was given the President’s
Distinguished Federal Civilian Service Award by President John F. Kennedy, the
highest civilian honor that can be conferred on a government employee. The major
change in drug legislation caused by the thalidomide disaster was induced
by an increased public awareness and demand for drug safety. This
public need was the motivation for the Kefauver bill to be redrafted. The
revised Kefauver–Harris Amendment was signed into law by President Kennedy on
October 10, 1962. One of the most significant effects of this legislation was
the requirement that drugs were to be proven effective before they could be
marketed in the United States. A safety issue was transformed into a requirement
for proof of efficacy. It was the second major change in drug legislation and it
was enacted almost as an afterthought.
Proof of efficacy
was defi ned as the requirement that two adequate and
well-controlled studies confirm appropriate activity. This language, crafted in
the early 1960s, was to haunt the drug approval process for the next 35 years.
The dye was cast for drug development. The Elixir Sulfanilamide disaster led to
the requirement for proof of safety, and the thalidomide disaster was the
vehicle which ensured that a drug has to be proved effective before it can be
marketed. Everything relating to the drug approval process as we know it today
relates back to the requirement to prove safety and efficacy. The FDA had been
transformed from an agency that responded to negative drug issues to an agency
that proactively scrutinized new drug development.
Since the adoption of the Kefauver–Harris Amendment, there have
been innumerable modifications, additions, and changes made to the remit and
responsibilities of the FDA. For example, clinical research has had to respond
to the challenges of the global market. Different countries in the developed
world required different data to obtain marketing approvals. This led to slower,
more expensive development programs. It was proposed that within reasonable
limits, a safe and effective drug should not require vastly different clinical
development programs to gain approvals to be marketed in different parts of the
world. Europe had pioneered harmonization in the European Community in the 1980s
and initiated discussions with Japan and the United States on the harmonization
of drug development requirements. These discussions culminated in the birth of
the International Conference on Harmonization of Technical Requirements for
Registration of Pharmaceuticals for Human Use (ICH). This occurred at a meeting
of the European Federation of Pharmaceutical Industries and Associations in
April 1990 in Brussels. The ICH comprises representatives of the regulatory
bodies and research-based industry from Europe, Japan, and the United States.
The terms of reference for the ICH were agreed on and the topics
selected for harmonization were safety, quality, and efficacy. The focus was on
harmonizing the technical content of blocks of data where it was apparent that
there were significant regional variations in requirements. This resulted in
over 60 guidelines and revisions being published in the first 10 years that the
ICH existed. This phase of development was deemed complete at the 5th
International Conference on Harmonization (ICH 5) held in San Diego in November
2000. The main focus of the ICH switched to harmonization of the format and
content of registration applications. Ultimately, this will result in a common
technical document. Other areas targeted for harmonization include new
technological advances, new innovative medicines, and postmarketing issues. The
U.S. drug regulatory process is now more closely linked to Europe and Japan than
ever before. The ICH guidelines are implemented after publication in the
Federal Register. Fundamentally,
however, with regards to drugs, the societal mandate in the United States has
remained the same: The drug must be proven safe and effective. This led to the
development of arguably the most important piece of documentation in drug
development in the United States, the
new drug application
(NDA). If that document passes
the FDA approval process, the drug is deemed safe and effective within the scope
of the clinical program and may be given to patients. The content and format of
the NDA will continue to evolve, but
the basic tenets have endured: Ensure full comprehension of the
participant as to the risks of the study and its methods of eliciting usable
results, establish safety and efficacy, and going full circle historically,
return to the Hippocratic Oath, keep [subjects] from harm and injustice. Ethics
and development have come together through tortuous paths.
Preclinical Development
The aim of a clinical development program is to generate
sufficient data to satisfy the regulatory requirements for allowing the drug to
be marketed. The endpoint of this process is
to prove that the drug is safe and effective in humans. The
doorway to testing the drug in humans in the United States is to generate an
investigational new drug
(IND) application. The initial animal studies to
determine pharmacological effects are usually conducted using laboratory-scale
drug synthesis. After the initial in vitro and in vivo tests have shown
preclinical “proof of principle”, the scale-up process is begun. The initial
scale-up is usually between a few hundred grams to a kilogram, depending on the
complexity of the synthesis and whether the synthetic route is scalable (i.e.,
chromatography steps can be accommodated in the scale-up or there are no
potentially explosive steps that would preclude scale-up). For drugs that have
little or no toxicity, the scale-up will have to be on the order of tens of
kilograms, as the IND enabling toxicology evaluations may have to go to 100-fold
the expected human dose. The next major event is the manufacture of good
manufacturing practice (GMP) material.
Good manufacturing practice
is a system for ensuring that products are produced
consistently and controlled according to quality standards. This system forms an
integral part of the manufacturing process. It is considered necessary for the
following reasons:
• To assure consistency between and within batches of the
investigational product and thus assure the reliability of clinical trials
• To assure consistency between the investigational product and
the future commercial product and therefore the relevance of the clinical trial
to the efficacy and safety of the marketed product
• To protect subjects of clinical trials from poor-quality
products resulting from manufacturing
errors (omission of critical steps such as sterilization,
contamination and cross-contamination, mix-ups, wrong labeling, etc.) or from
starting materials and components of inadequate quality
• To document all changes in the manufacturing process, in the
early clinical trials the dosage form may be different from the commercial
product (e.g., capsule instead of tablet), but for the pivotal studies on which
registration is based it should be the same as for the commercialized product.
It is accepted that validated analytical procedures may not always be available
for the early clinical trials, so provisional production parameters and
in-process controls should be deduced from experience with analogous products.
In short, the GMP system evolves during the clinical development but should be
the same as is required by marketed product for the drug that is used in the
pivotal trials.
It is not necessary to manufacture material for any of the
preclinical studies to GMP. Some companies like to use GMP material for the
IND-enabling toxicology studies, but a reasonable compromise is to use material
that has a certificate of analysis. The progression in GMP manufacture is
usually facilitated if the last non-GMP batch is larger than the GMP batch. The
latter faces only the stringency of the in-process controls and intermediate and
API release specifications, not the potential problems of those controls applied
to a scale that has not previously been manufactured. The aim in the succeeding
scale-up is not to produce a drug that is significantly different (API or
impurities) from the drug that was used in the IND-enabling in vitro and in vivo
studies.
There are two main reasons for conducting preclinical in vitro
and in vivo studies:
• To characterize the drug and investigate its utility in models
of possible therapeutic targets
• To satisfy the regulatory requirements to allow clinical
development of the drug to advance
The characterization and assessment of possible utility are not
subject to any stringent controls other than ethical committee approval when
animals are used. As an example of such a program, here is a potential
feasibility assessment of a topical application of a drug to treat atopic
dermatitis. The initial study could be to determine activity: for example,
applying the drug topically to prevent an injection of zymosan-inducing paw
edema in a mouse model, or topical application to prevent a delayed-type
hypersensitivity response to 2,4-dinitrofl urobenzene in a mouse model. The next
stage in characterization could be assessing penetration of the drug to the
dermis, which is the target site. The mini-pig has skin which is structurally
similar to human skin, so that an analysis of a cutaneous punch biopsy after
topical application of the drug would determine penetration. The third model in
the proof of principle program would be topical application of the drug to a
mini-pig model of atopic dermatitis to assess efficacy. In three simple studies,
answers are found as to whether the drug delivered topically is active,
penetrates to the required site of action, and is effective. It must be accepted
that animal models are not necessarily predictive of human efficacy or safety
(e.g., the failure of animal models to predict the mutagenic activity of
thalidomide), but a positive set of efficacy results in animal studies is
usually grounds for advancing the development program. Should the results of the
animal studies be negative, there is scant reason to continue the program. These
studies are not usually subjected to the preclinical documentation requirement
of
good laboratory practice
(GLP), the equivalent for preclinical testing to GMP
for manufacturing. However, these studies, if conducted pre-IND, will be used in
the pharmacology/toxicology section of the IND to establish a rationale for
exposing the drug to humans and must therefore be adequately documented.
In the same way as the bureaucratic noose tightens the GMP
requirement as the development program approaches the pivotal clinical studies,
so the bureaucratic requirements for preclinical testing strengthen as the
studies become the determinant of whether a drug can be given to humans.
Throughout the clinical development, additional nonclinical studies are
required, but initially the requirement is for IND enabling studies so that the
drug can be given to humans. The standard requirements are:
1. Single- and repeat-dose toxicity studies
2. Pharmacokinetic, toxicokinetic, and absorption, distribution,
metabolism, and
excretion (ADME) studies
3. Genotoxicity studies
4. Safety pharmacology studies
It should be noted that all of the studies listed above relate to
drug safety, and none to preclinical proof of concept. This is the dividing line
between studies that require the bureaucratic vigor of GLP and those that do
not. The IND-enabling studies have to be conducted to GLP standards. GLP was
developed to promote the quality and validity of the test data used for
determining the safety of chemicals and chemical products. The FDA published GLP
regulations for nonclinical studies in 1976, and they provided the basis of the
Organisation for Economic Co-operation and Development guidelines that made GLP
international in 1978. Like the Declaration of Helsinki and the ICH guidelines,
the OECD GLP guidelines remain under continuous review and are updated
periodically.
Good laboratory practice is a quality system concerned with the
organizational process and the conditions under which nonclinical health and
environmental safety studies are planned, performed, monitored, archived, and
reported. The guidelines examine the requirements for test facility organization
and personnel, such as qualifications, training (including training logs of
updates), and standard operating procedures. They defi ne the requirements of
the quality assurance program, including documentation, inspections, and
sign-off of final reports. There are guidelines on facility utilization,
apparatus, materials and reagents, test systems, test articles, study
performance, report quality, and the storage and retention of records and
materials.
1.
Toxicity studies. One program in the safety evaluation straddles the
requirement for GLP. The toxicology program frequently starts with dose-finding
studies to determine the high, middle, and low doses that will be used in the
repeat-dose toxicity studies to support phase I clinical studies. These studies
can include single-dose escalation and short-duration multiple-dose studies
(approximately 5-day dosing). Usually, these studies record gross pathology
without histology. They are not subject to GLP but will, of course, be reported
in the IND. The IND-enabling repeat-dose toxicity studies should be conducted
under GLP. These studies are usually conducted in two mammalian species (one
nonrodent) and should be equal or exceed the duration of the human clinical
trials proposed. If, in the later stages of development, the duration of the
dosing period increases in the human clinical trials, additional toxicity
studies must be conducted of sufficient duration to support those trials. For a
chronic treatment it is necessary to conduct a six-month study in rodents and at
leasta nine-month study in nonrodents.
2.
Pharmacokinetic, toxicokinetic,
and ADME studies.
As a part of the toxicity studies, or in other studies, toxicokinetics should be
performed.
Toxicokinetics
is defi ned as the generation of pharmacokinetic data
in order to assess systemic exposure. These data can be used in the
interpretation of toxicology fi ndings and their relevance to clinical safety.
For meaningful results to be generated, analytical methods must have been
developed with the analytes (API, metabolites, etc.) and matrices (plasma, whole
blood, tissue, etc.). These methods are under continuous review as additional
information is gathered on metabolism and species differences. The drug and
metabolite distribution in tissues should be determined. The ICH members are in
agreement that single-dose distribution studies should form part of the
preclinical evaluation. There are circumstances when it will be necessary to
conduct repeat-dose tissue distribution studies. They would be appropriate for
compounds that have a long half-life, incomplete elimination, or unanticipated
organ toxicity.
3.
Genotoxicity. Tests are designed to show whether a drug can
induce genetic damage. The ICH guidance on the standard battery for genotoxicity
testing of drugs advocates the following as an initial assessment:
• A test for gene mutation in bacteria
• An in vitro test of chromosomal damage
• An in vivo test of chromosomal damage using rodent
hematopoietic cells
If these tests are negative, it is usually considered that no
additional testing of genotoxic activity is required. Positive tests will
require additional evaluations.
4.
Safety pharmacology studies. Safety pharmacology studies are those studies that
determine undesirable pharmacodynamic effects of a drug on physiological
functions. The most important of these are effects of physiological functions
that are critical for life, cardiovascular, respiratory, and central nervous
systems. However, if a drug is targeted to affect a disease process in another
system with a specific effect such inflammatory cell migration in the
gastrointestinal tract, closer scrutiny of that system may be necessary.
Hot topic issues will always ensure that this preclinical
category remains under consideration.
Recent acknowledgment of enhanced proarrhythmic risk is an
example. Much attention is being paid to nonclinical evaluation of the potential
for delayed repolarization (QT interval prolongation) by pharmaceuticals. Like
the process chemistry modifications and formulation development, the nonclinical
evaluation process continues well past the IND enabling phase. Additional
preclinical evaluation will be necessary. For example, if human beings of
reproductive age will be treated by the drug, reproductive toxicology will be
necessary before an NDA can be fi led, and if the drug is to be dosed
chronically, carcinogenicity studies will be required. The nonclinical
evaluation process continues throughout the clinical development. Once suffi
cient preclinical data have been gathered, there is an opportunity to meet with
the FDA for a prefi ling IND assessment. This pre-IND meeting allows the sponsor
to outline the basic elements that will be in the IND and to seek the FDA’s view
as to whether the filing appears to meet the requirements that will allow the
IND to be approved and thus allow clinical trials to begin. The structure of the
IND is detailed below.
1.
Introductory statement. This includes the name of the drug and all active
ingredients, the pharmacological class, the structural formula, the method of
formulation, the route of administration, and a summary of previous human
experience.
2.
General investigational plan. This should cover the investigations that will be
conducted during the next year and a rationale for this approach.
3.
Investigator’s brochure. The most important feature of this document, which
is the primer for the investigating physician, is the summary of safety and
efficacy and the pharmacokinetics
and biological distribution of the drug in animals.
4.
Protocols. These give the details of the types of studies that
will be conducted, in which subject population, for how long, with what
variables. The protocols also list the qualifications of the investigators and
subinvestigators and the name and address of the investigational review board.
5.
Chemistry, manufacturing, and
control
(CMC)
information
a. Drug substance [active pharmaceutical ingredient (API)],
including the general methods of preparation, the analytical methods used to
assure identity, strength, quality, and purity, and data supporting the
stability of the drug substance for the duration of the toxicology studies.
b. Drug product, including all components, analytical methods for
release, a brief description of the manufacturing and packaging procedures, and
sufficient data to assure the product’s stability during the planned clinical
studies.
6.
Pharmacology and toxicology
(pharm/tox) information.
This includes data from animal
and in vitro studies:
a. Pharmacological effects and mechanisms of action and drug
disposition, including effects and mechanisms of action and information on
absorption, distribution, metabolism, and excretion. Safety pharmacology data
must be available on the effects in animals on vital functions such as
cardiovascular, central nervous, and respiratory systems.
b. Integrated summary of toxicological effects of the drug in
animals and in vitro. The clinical studies proposed determine the duration of
toxicology testing required, whether reproductive toxicology is required, and
whether special toxicity tests due to the drug’s route of administration are
required. Prior to human exposure, in vitro tests for the evaluation of
mutations and chromosomal damage are generally required.
There are a number of other sections that may be required, depending on
the drug being evaluated. These include sections on previous human experience
and dependence and abuse potential.
The data-driven elements in the IND are the CMC and pharm/tox
Sections. The mechanism of drug synthesis has to have been defi ned and
analytical methods developed to ensure reproducible quality that is sustainable
over time. It is recognized that as manufacturing scale-up occurs, changes will
take place in the synthetic pathway, but care has to be taken to ensure that the
drug substance and impurity profile remain the same as for the batches of drug
substance used in the IND enabling toxicology or safety pharmacology studies. If
there is a significant deviation from the analytical release specifications, it
could invalidate the results of those studies, which would have to be repeated;
bridging studies conducted to show that the current drug is essentially similar
in its effects to the original drug; or in the worse-case scenario, a new IND
would have to be filed.
The fi ling of an IND takes the development process to the next
level. If the IND is accepted by the FDA [i.e., if it conforms to the content
and format laid down in the Code of Federal Regulations (21 CFR 312.23)], the
FDA has 30 days to comment. At the end of this time, the clinical study may be
started whether or not comments have been received. If the FDA determines that
there is inadequate information to justify administering the drug to humans, the
program is put on clinical hold until such times as adequate data are provided
to support giving the drug to humans, once this is achieved, the clinical phase
of the development program has begun.
Clinical research design
Clinical research design has two major types that include
non-interventional/observational and interventional/experimental studies. The
non-interventional studies may have a comparator group (analytical studies like
case-control and cohort studies), or without it (descriptive study). The
experimental studies may be either randomized or non-randomized. Clinical trial
designs are of several types that include parallel design, crossover design,
factorial design, randomized withdrawal approach, adaptive design, superiority
design, and non-inferiority design. The advantages and disadvantages of clinical
trial designs are depicted in Table.
Table - Clinical trial designs, their advantages, and disadvantages
|
Trial design type |
Type of the study |
Nature of study |
Advantages/disadvantages |
|
Parallel |
Randomized |
This is the most frequent design wherein each arm of the study group is
allocated a particular treatment (placebo (an inert
substance)/therapeutic drug) |
The placebo arm does not receive the trial drug, so may not get the
benefit of it |
|
Crossover |
Randomized |
The patient in this trial gets each drug and the
patients serve as a control themselves |
Avoids participant bias in treatment and requires
a small sample size. This design is not suitable for research on acute
diseases. |
|
Factorial |
Non-randomized |
Two or more interventions on the participants and the study can provide
information on the interactions between the drugs |
The study design is complex |
|
Randomized withdrawal approach |
Randomized |
This study evaluates the time/duration of the drug
therapy |
The study uses a placebo to understand the
efficacy of a drug in treating the disease |
|
Matched pairs |
Post-approval study |
Recruit patients with the same characteristics |
Less variability |
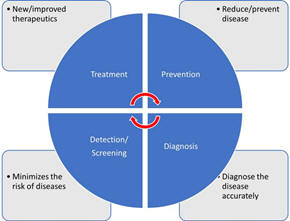
There are different types of clinical trials that include those which
are conducted for treatment, prevention, early detection/screening, and
diagnosis. These studies address the activities of an investigational drug on a
disease and its outcomes. They assess whether the drug is able to prevent the
disease/condition, the ability of a device to detect/screen the disease, and the
efficacy of a medical test to diagnose the disease/condition. The pictorial
representation of a disease diagnosis, treatment, and prevention is depicted in
Figure.
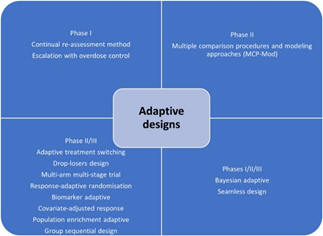
The Bayesian adaptive trial design has gained popularity, especially
during the Coronavirus Disease-19 (COVID-19) pandemic. Such designs could
operate under a single master protocol. It operates as a platform trial wherein
multiple treatments can be tested on different patient groups suffering from
disease.
A clinical trial involves the study of the effect of an
investigational drug/any other intervention in a defined population/participant.
The clinical research includes a treatment group and a placebo wherein each
group is evaluated for the efficacy of the intervention (improved/not improved).
Clinical trials are broadly classified into controlled and uncontrolled trials.
The uncontrolled trials are potentially biased, and the results of such research
are not considered as equally as the controlled studies. Randomized controlled
trials (RCTs) are considered the most effective clinical trials wherein the bias
is minimized, and the results are considered reliable. There are different types
of randomizations and each one has clearly defined functions as elaborated in
Table.
Table - Different types of randomizations in clinical trials
|
Randomization type |
Functions |
|
Simple randomization |
The participants are assigned to a case or a control group based on
flipping coin results/computer assignment |
|
Block randomization |
Equal and small groups of both cases and controls |
|
Stratified randomization |
Randomization based on the age of the participant and other covariates |
|
Co-variate adaptive randomization/minimization |
Sequential assignment of a new participant into a
group based on the covariates |
|
Randomization by body halves or paired organs (Split body trials) |
One intervention is administered to one-half of the body and the
comparator intervention is assigned to another half of the body |
|
Clustered randomization |
Intervention is administered to clusters/groups by
randomization to prevent contamination and either active or comparator
intervention is administered for each group |
|
Allocation by randomized consent (Zelen trials) |
Patients are allocated to one of the two trial arms |
Principles of clinical trial/research
Clinical trials or clinical research are conducted to improve the
understanding of the unknown, test a hypothesis, and perform public
health-related research. This is majorly carried out by collecting the data and
analyzing it to derive conclusions. There are various types of clinical trials
that are majorly grouped as analytical, observational, and experimental
research. Clinical research can also be classified into non-directed data
capture, directed data capture, and drug trials. Clinical research could be
prospective or retrospective. It may also be a case-control study or a cohort
study. Clinical trials may be initiated to find treatment, prevent, observe, and
diagnose a disease or a medical condition.
Among the various types of clinical research, observational research using a
cross-sectional study design is the most frequently performed clinical research.
This type of research is undertaken to analyze the presence or absence of a
disease/condition, potential risk factors, and prevalence and incidence rates in
a defined population. Clinical trials may be therapeutic or non-therapeutic type
depending on the type of intervention. The therapeutic type of clinical trial
uses a drug that may be beneficial to the patient. Whereas in a non-therapeutic
clinical trial, the participant does not benefit from the drug. The
non-therapeutic trials provide additional knowledge of the drug for future
improvements. Different terminologies of clinical trials are delineated in
Table.
Table - Clinical trial methods and terminologies
|
Type of clinical trial |
Definition |
|
Randomized trial |
Study participants are randomly assigned to a group |
|
Open-label |
Both study subjects and the researchers are aware
of the drug being tested |
|
Blinded (single-blind) |
In single-blind studies, the subject has no idea about the group
(test/control) in which they are placed |
|
Double-blind (double-blind) |
In the double-blind study, the subjects as well as
the investigator have no idea about the test/control group |
|
Placebo |
A substance that appears like a drug but has no active moiety |
|
Add-on |
An additional drug apart from the clinical trial
drug given to a group of study participants |
|
Single center |
A study being carried out at a particular place/location/center |
|
Multi-center |
A study is being carried out at multiple
places/locations/centers |
In view of the increased
cost of the drug discovery process, developing, and low-income countries
depend on the production of generic drugs. The generic drugs are similar in
composition to the patented/branded drug. Once the patent period is expired
generic drugs can be manufactured which have a similar quality, strength, and
safety as the patented drug. The regulatory requirements and the drug production
process are almost the same for the branded and the generic drug according to
the Food and Drug Administration (FDA), United States of America (USA).
The bioequivalence (BE) studies review the absorption, distribution,
metabolism, and excretion (ADME) of the generic drug. These studies compare the
concentration of the drug at the desired location in the human body, called the
peak concentration of the drug (Cmax). The extent of absorption of the drug is
measured using the area under the receiver operating characteristic curve (AUC),
wherein the generic drug is supposed to demonstrate similar ADME activities as
the branded drug. The BE studies may be undertaken in vitro (fasting,
non-fasting, sprinkled fasting) or in vivo studies (clinical, bioanalytical, and
statistical).
Planning clinical trial/research
The clinical trial process involves protocol development, designing a case
record/report form (CRF), and functioning of institutional review boards (IRBs).
It also includes data management and the monitoring of clinical trial site
activities. The CRF is the most significant document in a clinical study. It
contains the information collected by the investigator about each subject
participating in a clinical study/trial. According to the International Council
for Harmonisation (ICH), the CRF can be printed, optical, or an electronic
document that is used to record the safety and efficacy of the pharmaceutical
drug/product in the test subjects. This information is intended for the sponsor
who initiates the clinical study.
The CRF is designed as per the protocol and later it is thoroughly reviewed
for its correctness (appropriate and structured questions) and finalized. The
CRF then proceeds toward the print taking the language of the participating
subjects into consideration. Once the CRF is printed, it is distributed to the
investigation sites where it is filled with the details of the participating
subjects by the investigator/nurse/subject/guardian of the
subject/technician/consultant/ monitors/pharmacist/pharmacokinetics/contract
house staff. The filled CRFs are checked for their completeness and transported
to the sponsor.
Effective planning and implementation of a clinical study/trial will
influence its success. The clinical study majorly includes the collection and
distribution of the trial data, which is done by the clinical data management
section. The project manager is crucial to effectively plan, organize, and use
the best processes to control and monitor the clinical study.
The clinical study is conducted by a sponsor or a clinical research
organization (CRO). A perfect protocol, time limits, and regulatory requirements
assume significance while planning a clinical trial. What, when, how, and who
are clearly planned before the initiation of a study trial. Regular review of
the project using the bar and Gantt charts, and maintaining the timelines assume
increased significance for success with the product (study report, statistical
report, database).
The steps critical to planning a clinical trial include the idea, review of
the available literature, identifying a problem, formulating the hypothesis,
writing a synopsis, identifying the investigators, writing a protocol, finding a
source of funding, designing a patient consent form, forming ethics boards,
identifying an organization, preparing manuals for procedures, quality
assurance, investigator training and initiation of the trial by recruiting the
participants. The two most important points to consider before the initiation of
the clinical trial include whether there is a need for a clinical trial, if
there is a need, then one must make sure that the study design and methodology
are strong for the results to be reliable to the people.
For clinical research to envisage high-quality results, the study design,
implementation of the study, quality assurance in data collection, and
alleviation of bias and confounding factors must be robust. Another important
aspect of conducting a clinical trial is improved management of various elements
of clinical research that include human and financial resources. The role of a
trial manager to make a successful clinical trial was previously reported. The
trial manager could play a key role in planning, coordinating, and successfully
executing the trial. Some qualities of a trial manager include better
communication and motivation, leadership, and strategic, tactical, and
operational skills.
Practical aspects of a clinical trial operations
There are different types of clinical research. Research in the development
of a novel drug could be initiated by nationally funded research,
industry-sponsored research, and clinical research initiated by
individuals/investigators. According to the documents 21 code of federal
regulations (CFR) 312.3 and ICH E-6 Good Clinical Practice (GCP) 1.54, an
investigator is an individual who initiates and conducts clinical research. The
investigator plan, design, conduct, monitor, manage data, compile reports, and
supervise research-related regulatory and ethical issues. To manage a successful
clinical trial project, it is essential for an investigator to give the letter
of intent, write a proposal, set a timeline, develop a protocol and related
documents like the case record forms, define the budget, and identify the
funding sources.
Other major steps of clinical research include the approval of IRBs,
conduction and supervision of the research, data review, and analysis.
Successful clinical research includes various essential elements like a letter
of intent which is the evidence that supports the interest of the researcher to
conduct drug research, timeline, funding source, supplier, and participant
characters. Quality assurance, according to the ICH and GCP guidelines, is
necessary to be implemented during clinical research to generate quality and
accurate data. Each element of the clinical research must have been carried out
according to the standard operating procedure (SOP), which is written/determined
before the initiation of the study and during the preparation of the protocol.
The audit team (quality assurance group) is instrumental in determining the
authenticity of the clinical research. The audit, according to the ICH and GCP,
is an independent and external team that examines the process (recording the
CRF, analysis of data, and interpretation of data) of clinical research. The
quality assurance personnel are adequately trained, become trainers if needed,
should be good communicators, and must handle any kind of situation. The audits
can be at the investigator sites evaluating the CRF data, the protocol, and the
personnel involved in clinical research (source data verification,
monitors).Clinical trial operations are governed by legal and regulatory
requirements, based on GCPs, and the application of science, technology, and
interpersonal skills. Clinical trial operations are complex, time and
resource-specific that requires extensive planning and coordination, especially
for the research which is conducted at multiple trial centers.
Recruiting the clinical trial participants/subjects is the most significant
aspect of clinical trial operations. Previous research had noted that most
clinical trials do not meet the participant numbers as decided in the protocol.
Therefore, it is important to identify the potential barriers to patient
recruitment. Most clinical trials
demand huge costs, increased timelines, and resources. Randomized clinical trial
studies from Switzerland were analyzed for their costs which revealed
approximately 72000 USD for a clinical trial to be completed. This study
emphasized the need for increased transparency with respect to the costs
associated with the clinical trial and improved collaboration between
collaborators and stakeholders.
Clinical trial applications, monitoring, and audit
Among the most significant aspects of a clinical trial is the audit. An audit
is a systematic process of evaluating the clinical trial operations at the site.
The audit ensures that the clinical trial process is conducted according to the
protocol, and predefined quality system procedures, following GCP guidelines,
and according to the requirements of regulatory authorities. The auditors are
supposed to be independent and work without the involvement of the sponsors,
CROs, or personnel at the trial site. The auditors ensure that the trial is
conducted by designated professionally qualified, adequately trained personnel,
with predefined responsibilities. The auditors also ensure the validity of the
investigational drug, and the composition, and functioning of institutional
review/ethics committees. The availability and correctness of the documents like
the investigational broacher, informed consent forms, CRFs, approval letters of
the regulatory authorities, and accreditation of the trial labs/sites.
The data management systems, the data collection software, data backup,
recovery, and contingency plans, alternative data recording methods, security of
the data, personnel training in data entry, and the statistical methods used to
analyze the results of the trial are other important responsibilities of the
auditor. According to the
ICH-GCP Sec 1.29 guidelines the inspection may be described as an act by the
regulatory authorities to conduct an official review of the clinical
trial-related documents, personnel (sponsor, investigator), and the trial site.
The summary report of the observations of the inspectors is performed using
various forms as listed in Table.
Table - The FDA regulatory forms for the submission of inspection results
FDA: Food and Drug Administration; IND: investigational new drug; NDA: new drug
application; IRB: institutional review board; CFR: code of federal regulations
|
Regulatory (FDA) form
number |
Components of the form |
|
483 |
List of objectionable conditions/processes prepared by the FDA
investigator and submitted to the auditee at the end of the inspection |
|
482 |
The auditors submit their identity proofs and
notice of inspections to the clinical investigators and later document
their observations |
|
1571 |
This document details the fact that the clinical trial is not initiated
before 30 days of submitting the IND to the FDA for approval. The form
confirms that the IRB complies with 21 CFR Part 56. The form details the
agreement to follow regulatory requirements and names all the
individuals who monitor the conduct and progress of the study and
evaluate the safety of the clinical trial |
|
1572 |
This form details the fact that the study is
conducted after ethics approval ensures that the study is carried out
according to protocol, informed consent, and IR |
Because protecting data integrity, the rights, safety, and well-being of the
study participants are more significant while conducting a clinical trial,
regular monitoring and audit of the process appear crucial. Also, the quality of
the clinical trial greatly depends on the approach of the trial personnel which
includes the sponsors and investigators.
The responsibility of monitoring lies in different hands, and it depends
on the clinical trial site. When the trial is initiated by a pharmaceutical
industry, the responsibility of trial monitoring depends on the company or the
sponsor, and when the trial is conducted by an academic organization, the
responsibility lies with the principal investigator.
An audit is a process conducted by an independent body to ensure the quality
of the study. Basically, an audit is a quality assurance process that determines
if a study is carried out by following the SPOs, in compliance with the GCPs
recommended by regulatory bodies like the ICH, FDA, and other local bodies.
An audit is performed to review all the available documents related to
the IRB approval, investigational drug, and the documents related to the patient
care/case record forms. Other documents that are audited include the protocol
(date, sign, treatment, compliance), informed consent form, treatment
response/outcome, toxic response/adverse event recording, and the accuracy of
data entry.
Clinical trial data analysis, regulatory audits, and project management
The essential elements of clinical trial management systems (CDMS) include
the management of the study, the site, staff, subject, contracts, data, and
document management, patient diary integration, medical coding, monitoring,
adverse event reporting, supplier management, lab data, external interfaces, and
randomization. The CDMS involves setting a defined start and finishing time,
defining study objectives, setting enrolment and termination criteria,
commenting, and managing the study design.
Among the various key application areas of clinical trial systems, the
data analysis assumes increased significance. The clinical trial data collected
at the site in the form of case record form is stored in the CDMS ensuring the
errors with respect to the double data entry are minimized.
Table -Types of clinical trial audits
CRF: case report form; CSR: clinical study report; IC: informed consent; PV:
pharmacovigilance; SAE: serious adverse event
|
Product-specific audits program |
Pharmacovigilance audits program |
|
Protocol, CRF, IC, CSR |
|
|
Supplier |
Safety data management |
|
Clinical database |
|
|
Investigator site |
Communications and regulatory reporting |
|
Clinical site visit |
|
|
Study management |
Signal detection and evaluation |
|
SAE reporting |
|
|
Supplier audits program |
Risk management and PV planning |
|
Supplier qualification |
|
|
Sponsor data audit during the trial |
Computerized system |
|
Preferred vendor list after the trials |
|
|
Process/System audits program |
Suppliers |
|
Clinical safety reporting |
|
|
Data management |
Regulatory inspection management program |
|
Clinical supply |
|
|
Study monitoring |
Assist with the audit response |
|
Computerized system |
Pre-inspection audit |
Clinical trial data management uses medical coding, which uses terminologies
with respect to the medications and adverse events/serious adverse events that
need to be entered into the CDMS. The project undertaken to conduct the clinical
trial must be predetermined with timelines and milestones. Timelines are usually
set for the preparation of protocol, designing the CRF, planning the project,
identifying the first subject, and timelines for recording the patient’s data
for the first visit. The timelines also are set for the last subject to be
recruited in the study, the CRF of the last subject, and the locked period after
the last subject entry. The planning of the project also includes the modes of
collection of the data, the methods of the transport of the CRFs, patient
diaries, and records of severe adverse events, to the central data management
sites (fax, scan, courier, etc.).
The preparation of SOPs and the type and timing of the quality control (QC)
procedures are also included in the project planning before the start of a
clinical study. Review (budget, resources, quality of process, assessment),
measure (turnaround times, training issues), and control (CRF collection and
delivery, incentives, revising the process) are the three important aspects of
the implementation of a clinical research project.
In view of the increasing complexity related to the conduct of clinical
trials, it is important to perform a clinical quality assurance (CQA) audit. The
CQA audit process consists of a detailed plan for conducting audits, points of
improvement, generating meaningful audit results, verifying SOP, and regulatory
compliance, and promoting improvement in clinical trial research. All the
components of a CQA audit are delineated in Table.
Clinical trial operations at the investigator's site
The selection of an investigation site is important before starting a
clinical trial. It is essential that the individuals recruited for the study
meet the inclusion criteria of the trial, and the investigator's and
patient's willingness to accept the protocol design and the timelines set by the
regulatory authorities including the IRBs.
Before conducting clinical research, it is important for an investigator
to agree to the terms and conditions of the agreement and maintain the
confidentiality of the protocol. Evaluation of the protocol for the feasibility
of its practices with respect to the resources, infrastructure, qualified and
trained personnel available, availability of the study subjects, and benefit to
the institution and the investigator is done by the sponsor during the site
selection visit. The standards of a clinical research trial are ensured by the
Council for International Organizations of Medical Sciences (CIOMS), National
Bioethics Advisory Commission (NBAC), United Nations Programme on Human
Immunodeficiency Virus/Acquired Immunodeficiency Syndrome (HIV/AIDS) (UNAIDS),
and World Medical Association (WMA).
Recommendations for conducting clinical research based on the WMA support the
slogan that says, “The health of my patient will be my first
consideration.” According to the International Code of Medical Ethics (ICME), no
human should be physically or mentally harmed during the clinical trial, and the
study should be conducted in the best interest of the person. Basic principles
recommended by the Helsinki declaration include the conduction of clinical
research only after the prior proof of the safety of the drug in animal and lab
experiments. The clinical trials must be performed by scientifically, and
medically qualified and well-trained personnel. Also, it is important to analyze
the benefit of research over harm to the participants before initiating the drug
trials.
The doctors may prescribe a drug to alleviate the suffering of the patient,
save the patient from death, and gain additional knowledge of the drug only
after obtaining informed consent. Under the equipoise principle, the
investigators must be able to justify the treatment provided as a part of the
clinical trial, wherein the patient in the placebo arm may be harmed due to the
unavailability of the therapeutic/trial drug.
Clinical trial operations greatly depend on the environmental conditions and
geographical attributes of the trial site. It may influence the costs and
targets defined by the project before the initiation. It was noted that
one-fourth of the clinical trial project proposals/applications submit critical
data on the investigational drug from outside the country. Also, it was noted
that almost 35% of delays in clinical trials owing to patient recruitment with
one-third of studies enrolling only 5% of the participants.
It was suggested that clinical trial feasibility assessment in a defined
geographical region may be undertaken for improved chances of success. Points to
be considered under the feasibility assessment program include if the disease
under the study is related to the population of the geographical region,
appropriateness of the study design, patient, and comparator group, visit
intervals, potential regulatory and ethical challenges, and commitments of the
study partners, CROs in respective countries (multi-centric studies).
Feasibility assessments may be undertaken at the program level (ethics,
regulatory, and medical preparedness), study level (clinical, regulatory,
technical, and operational aspects), and at the investigation site
(investigational drug, competency of personnel, participant recruitment, and
retention, quality systems, and infrastructural aspects).
Clinical trials: true experiments
In accordance with the revised schedule "Y" of the Drugs and Cosmetics Act
(DCA) (2005), a drug trial may be defined as a systematic study of a novel drug
component. The clinical trials aim to evaluate the pharmacodynamic, and
pharmacokinetic properties including ADME, efficacy, and safety of new drugs.
According to the drug and cosmetic rules (DCR), 1945, a new chemical
entity (NCE) may be defined as a novel drug approved for a disease/condition, in
a specified route, and at a particular dosage. It also may be a new drug
combination, of previously approved drugs.
A clinical trial may be performed in three types; one that is done to
find the efficacy of an NCE, a comparison study of two drugs against a medical
condition, and the clinical research of approved drugs on a disease/condition.
Also, studies of the bioavailability and BE studies of the generic drugs, and
the drugs already approved in other countries are done to establish the efficacy
of new drugs.
Apart from the discovery of a novel drug, clinical trials are also conducted
to approve novel medical devices for public use. A medical device is defined as
any instrument, apparatus, appliance, software, and any other material used for
diagnostic/therapeutic purposes. The medical devices may be divided into three
classes wherein class I uses general controls; class II uses general and special
controls, and class III uses general, special controls, and premarket approvals.
The premarket approval applications ensure the safety and effectiveness,
and confirmation of the activities from bench to animal to human clinical
studies. The FDA approval for investigational device exemption (IDE) for a
device not approved for a new indication/disease/condition. There are two types
of IDE studies that include the feasibility study (basic safety and potential
effectiveness) and the pivotal study (trial endpoints, randomization,
monitoring, and statistical analysis plan).
As evidenced by the available literature, there are two types of research
that include observational and experimental research. Experimental research is
alternatively known as the true type of research wherein the research is
conducted by the intervention of a new drug/device/method (educational
research). Most true experiments use randomized control trials that remove bias
and neutralize the confounding variables that may interfere with the results of
research. The variables that may
interfere with the study results are independent variables also called
prediction variables (the intervention), dependent variables (the outcome), and
extraneous variables (other confounding factors that could influence the
outside). True experiments have three basic elements that include manipulation
(that influence independent variables), control (over extraneous influencers),
and randomization (unbiased grouping).
Experiments can also be grouped as true, quasi-experimental, and
non-experimental studies depending on the presence of specific characteristic
features. True experiments have all three elements of study design
(manipulation, control, randomization), and prospective, and have great
scientific validity. Quasi-experiments generally have two elements of design
(manipulation and control), are prospective, and have moderate scientific
validity. The non-experimental studies lack manipulation, control, and
randomization, are generally retrospective, and have low scientific validity.
Clinical trials: epidemiological and human genetics study
Epidemiological studies are intended to control health issues by
understanding the distribution, determinants, incidence, prevalence, and impact
on health among a defined population. Such studies are attempted to perceive the
status of infectious diseases as well as non-communicable diseases.
Experimental studies are of two types that include observational
(cross-sectional studies (surveys), case-control studies, and cohort studies)
and experimental studies (randomized control studies). Such research may pose
challenges related to ethics in relation to the social and cultural milieu.
Biomedical research related to human genetics and transplantation research
poses an increased threat to ethical concerns, especially after the success of
the human genome project (HGP) in the year 2000. The benefits of human genetic
studies are innumerable that include the identification of genetic diseases, in
vitro fertilization, and regeneration therapy. Research related to human
genetics poses ethical, legal, and social issues (ELSI) that need to be
appropriately addressed. Most importantly, these genetic research studies use
advanced technologies which should be equally available to both economically
well-placed and financially deprived people. Gene therapy and genetic
manipulations may potentially precipitate conflict of interest among the family
members. The research on genetics may be of various types that include pedigree
studies (identifying abnormal gene carriers), genetic screening (for diseases
that may be heritable by the children), gene therapeutics (gene replacement
therapy, gene construct administration), HGP (sequencing the whole human
genome/deoxyribonucleic acid (DNA) fingerprinting), and DNA, cell-line
banking/repository. The biobanks are established to collect and store human
tissue samples like umbilical tissue, cord blood, and others.
Epidemiological studies on genetics are attempts to understand the prevalence
of diseases that may be transmitted among families. The classical
epidemiological studies may include single case observations (one individual),
case series (< 10 individuals), ecological studies (population/large group of
people), cross-sectional studies (defined number of individuals), case-control
studies (defined number of individuals), cohort (defined number of individuals),
and interventional studies (defined number of individuals).
Genetic studies are of different types that include familial aggregation
(case-parent, case-parent-grandparent), heritability (study of twins),
segregation (pedigree study), linkage study (case-control), association,
linkage, disequilibrium, cohort case-only studies (related case-control,
unrelated case-control, exposure, non-exposure group, case group),
cross-sectional studies, association cohort (related case-control, familial
cohort), and experimental retrospective cohort (clinical trial, exposure, and
non-exposure group).
Ethics and concerns in clinical trial/research
Because clinical research involves animals and human participants, adhering
to ethics and ethical practices assumes increased significance. In view of the
unethical research conducted on war soldiers after the Second World War, the
Nuremberg code was introduced in 1947, which promulgated rules for permissible
medical experiments on humans. The Nuremberg code suggests that informed consent
is mandatory for all the participants in a clinical trial, and the study
subjects must be made aware of the nature, duration, and purpose of the study,
and potential health hazards (foreseen and unforeseen). The study subjects
should have the liberty to withdraw at any time during the trial and to choose a
physician upon medical emergency. The other essential principles of clinical
research involving human subjects as suggested by the Nuremberg code included
benefit to the society, justification of study as noted by the results of the
drug experiments on animals, avoiding even minimal suffering to the study
participants, and making sure that the participants don’t have life risk,
humanity first, improved medical facilities for participants, and suitably
qualified investigators.
During the 18th world medical assembly meeting in the year 1964, in Helsinki,
Finland, ethical principles for doctors practicing research were proposed.
Declaration of Helsinki, as it is known made sure that the interests and
concerns of the human participants will always prevail over the interests of the
society. Later in 1974, the National Research Act was proposed which made sure
that the research proposals are thoroughly screened by the Institutional
ethics/Review Board. In 1979, the April 18th Belmont report was proposed by the
national commission for the protection of human rights during biomedical and
behavioral research. The Belmont report proposed three core principles during
research involving human participants that include respect for persons,
beneficence, and justice. The ICH laid down GCP guidelines. These guidelines are
universally followed throughout the world during the conduction of clinical
research involving human participants.
ICH was first founded in 1991, in Brussels, under the umbrella of the USA,
Japan, and European countries. The ICH conference is conducted once every two
years with the participation from the member countries, observers from the
regulatory agencies, like the World Health Organization (WHO), European Free
Trade Association (EFTA), and the Canadian Health Protection Branch, and other
interested stakeholders from the academia and the industry. The expert working
groups of the ICH ensure the quality, efficacy, and safety of the medicinal
product (drug/device). Despite the availability of the Nuremberg code, the
Belmont Report, and the ICH-GCP guidelines, in the year 1982, International
Ethical Guidelines for Biomedical Research Involving Human Subjects was proposed
by the CIOMS in association with WHO. The CIOMS protects the rights of the
vulnerable population, and ensures ethical practices during clinical research,
especially in underdeveloped countries. In India, the ethical principles for
biomedical research involving human subjects were introduced by the Indian
Council of Medical Research (ICMR) in the year 2000 and were later amended in
the year 2006. Clinical trial approvals can only be done by the IRB approved by
the Drug Controller General of India (DGCI) as proposed in the year 2013.
Current perspectives and future implications
A recent study attempted to evaluate the efficacy of adaptive clinical trials
in predicting the success of a clinical trial drug that entered phase 3 and
minimizing the time and cost of drug development. This study highlighted the
drawbacks of such clinical trial designs that include the possibility of type 1
(false positive) and type 2 (false negative) errors.
The usefulness of animal studies during the preclinical phases of a
clinical trial was evaluated in a previous study which concluded that animal
studies may not completely guarantee the safety of the investigational drug.
This is noted by the fact that many drugs which passed toxicity tests in animals
produced adverse reactions in humans.
The significance of BE studies to compare branded and generic drugs was
reported previously. The pharmacokinetic BE studies of Amoxycillin comparing
branded and generic drugs were carried out among a group of healthy
participants. The study results have demonstrated that the generic drug had
lower Cmax as compared to the branded drug.
To establish the BE of the generic drugs, randomized crossover trials are
carried out to assess the Cmax and the AUC. The ratio of each pharmacokinetic
characteristic must match the ratio of AUC and/or Cmax, 1:1=1 for a generic drug
to be considered as a bioequivalent to a branded drug.
Although the generic drug development is comparatively more beneficial
than the branded drugs, synthesis of extended-release formulations of the
generic drug appears to be complex. Since the extended-release formulations
remain for longer periods in the stomach, they may be influenced by gastric
acidity and interact with the food. A recent study suggested the use of
bio-relevant dissolution tests to increase the successful production of generic
extended-release drug formulations.
Although RCTs are considered the best designs, which rule out bias and the
data/results obtained from such clinical research are the most reliable, RCTs
may be plagued by miscalculation of the treatment outcomes/bias, problems of
cointerventions, and contaminations.
The perception of healthcare providers regarding branded drugs and their view
about the generic equivalents was recently analyzed and reported. It was noted
that such a perception may be attributed to the flexible regulatory requirements
for the approval of a generic drug as compared to a branded drug. Also, could be
because a switch from a branded drug to a generic drug in patients may
precipitate adverse events as evidenced by previous reports.
Because the vulnerable population like drug/alcohol addicts, mentally
challenged people, children, geriatric age people, military persons, ethnic
minorities, people suffering from incurable diseases, students, employees, and
pregnant women cannot make decisions with respect to participating in a clinical
trial, ethical concerns, and legal issues may prop up, that may be appropriately
addressed before drug trials which include such groups.
Clinical Development
The IND is a living document that remains open for as long as the
drug remains under investigation. As each new study is designed, the sponsor
must submit to the FDA a protocol amendment containing the protocol for the next
study. Safety reports must be made to the FDA within 15 calendar days of the
sponsor being made aware of either a serious and unexpected adverse experience
or any fi nding from nonclinical studies which suggests a significant risk to
human subjects, such as reports of mutagenicity, teratogenicity, or
carcinogenicity. The sponsor must submit annual reports that contain individual
study information and summary information which comprises all information
obtained during the previous year’s clinical and nonclinical investigations,
including a summary of all safety reports generated during the year. Information
amendments are made to the IND when essential information is not contained in
any protocol amendment, safety report, or annual report. An example of the
content of an information amendment would be new chemistry or other technical
information. The clinical phase of the development program is the most complex
and costly. The overriding concern is for the safety of the clinical trial
subjects. Like the manufacturing code of practice (GMP), and the preclinical
evaluation code of practice (GLP), there is a code of practice for the human
investigation phase of the development program,
good clinical practice
(GCP). Unlike GMP and GLP, where there are
development stages that do not need to adhere to these regulations, all clinical
trials must conform to GCP. The ICH GCP guidance is broadly in line with the
Declaration of Helsinki. The main sections deal with:
• The Institutional Review Board
• The responsibilities of the sponsor of the clinical trial and
the investigator who will conduct the trial
• The clinical trial protocol
• The investigator’s brochure
• The essential documents for the conduct of a clinical trial
This aim of GCP guidance is to minimize the potential for harm to
befall a clinical trial subject. This governs the entire structure of the
clinical development program. For example, the numbers of patients entered into
studies would form an inverted pyramid. The earliest evaluations will be
conducted in less than 100 subjects, while the last studies in the clinical
development program may include hundreds or even thousands of subjects. This is
an attempt to minimize exposure to the drug at a stage when the least is known
about its possible toxicity and maximize it to determine safety and efficacy in
advance of applying for approval to market the drug. The goal of the clinical
development program is to generate sufficient clinical data of
acceptable quality to fi le a new drug application (NDA) with the
FDA so that they may determine whether the drug can be approved for marketing.
Probably the single most important
document in an NDA is the
label text, also called the
package insert, a summary of the drug that is made available to prescribers and
patients. The information that is contained in the label text summarizes what
the FDA has approved to be marketed. Any other use is called
off-label
and is not sanctioned by the FDA. If something goes wrong, the physician has no
defense to the accusation that he misused the drug. The marketing company is
also bound completely by the label text. There can be no advertising using
information outside the label text. The power of the document dictates that it
should be constructed before clinical development is started so that the
clinical research efforts are focused on that endpoint. When marketing, medical,
and regulatory departments first develop the label text there is often a
tendency to craft language that looks like: “tastes like chocolate, cures
cancer, and costs a buck.” As long as there is some justification based on the
nonclinical data for aiming at these best possible goals, they must be
considered feasible endpoints.
Of course, it must be accepted that this exercise will require
continual modification as hard data become available, but ultimately, by
focusing the clinical development program on the maximum potential of the drug,
the end result should be the best that the drug can deliver for the patients who
take it. If the preclinically devised label text is the esoteric goal of the
clinical development program, the operational aspect of the program is dictated
by need for clinical trial subject safety. The traditional terminology divides
the program into four temporally related phases. There are well-established
definitions of these phases:
Phase I
This phase starts with the first administration of the drug to
human subjects. The initial study in phase I (called phase Ia) is directed at an
evaluation of safety. As it often consists of single ascending doses, there may
be no opportunity to evaluate therapeutic efficacy endpoints. As the subjects
that are most often used in these studies are healthy volunteers, no
disease-related efficacy endpoint is possible. The subjects are usually male,
due to the requirement for reproduction toxicology data before women of
childbearing potential can be entered into clinical trials and there being no
necessity to submit such studies in the IND. The subjects in the majority of
phase I studies are admitted to hospital or a specialized phase I unit and
monitored continuously. The initial trial will use an open design; in other
words, both the investigator and the subject know that all doses will be test
articles and the doses of the test articles will be administered using an
ascending-dose regimen. A safety evaluation will be made after each dose is
administered before the next dose is given. The assessment of pharmacokinetics
and initial data on the characterization of the drug’s absorption, distribution,
metabolism, and excretion will be sought. There are too many exceptions to this
standard phase I evaluation of drugs to allow a comprehensive account of all
types of phase I programs. The following are common variants:
• The subjects are patients because the inherent toxicity of the
drug under evaluation precludes it from being given to volunteers; such a
situation may arise in the early evaluation of an anticancer drug.
• The subjects are patients because the disease process makes it
irrelevant what happens in volunteers; topically applied dermatological drugs
for the management of skin conditions are usually evaluated for safety in
patients that have a disrupted skin surface, as the healthy skin is a natural
barrier.
• A surrogate endpoint exists (e.g., a marker that shows activity
but is not predictive of therapeutic activity) so that pharmacodynamic data can
be generated. For example, a drug for asthma that exerted its effect by reducing
inflammatory mediator release from white cells had a pharmacodynamic evaluation
conducted in the initial phase I single-ascending-dose study. Peripheral white
cells were harvested and the release of the inflammatory mediator leukotreine B4
(LTB4) was monitored after inflammatory mediator release was stimulated by
calcium ionophore. The less LTB4 released after the stimulation, the better the
dose of the drug was working.
After the initial single-ascending-dose safety study is
completed, the next step is to conduct a multiple-dose safety study (phase Ib).
The aim in these studies is to get to steady state plasma concentrations (i.e.,
the highest concentrations that will ever be achieved form a particular dosage
regimen). The same potential variations in populations and endpoint exist in
this late phase I design as did in the first administration to human subjects in
the single-ascending-dose study. The multiple-dose study is often conducted
using an ascending-dose protocol where the lowest dose is given first and a
safety evaluation is conducted before administering the next-highest dose. In
cases where the nonclinical and phase Ia safety profiles are benign, it may be
possible to run the different doses in parallel, thereby saving a considerable
amount of development time. The total population required for the phase I
studies is on the order of 20 to 100. The industry norm for the duration of
phase I is less than one year. Of the drugs that enter phase I studies,
approximately 70% are progressed to the next phase.
Phase II
Once the drug has been shown to be safe, it will be tested for
efficacy. The population to be tested will be patients. Care is still taken to
minimize the risk to the study population. The inclusion and exclusion criteria
which defi ne patients that are eligible for the study usually exclude signifi
cant concomitant disease other than the disease or condition that the drug is
intended to prevent, diagnose, or treat. There are also usually restrictions on
the use of concomitant therapy. Considerable emphasis is still laid of safety
evaluation, but the patients may be treated as outpatients unless the disease
requires hospitalization. One of the most important endpoints of the phase II is
the definition of the dose or dose range to be taken into Phase III. The
objectives will include an assessment of the minimum dose that is maximally or
sufficiently effective and free of significant toxicity. An evaluation of the
primary endpoint to be used to determine efficacy in phase III is usually a
major objective.
For example, the results in nonclinical studies may suggest that
an investigational drug will accelerate the healing of a chronically painful
skin lesion and will be assessed in phase II/ with that endpoint. The proposed
label text will have language related to speed of healing. If, during phase II
it becomes apparent that healing is not accelerated but chronic pain is
controlled, the option will exist to advance to phase III, with the primary
variable being speed of healing in the hope that the phase II results were
anomalous, or switch to pain relief as the primary variable understanding that
the proposed label text will have to be changed to reflect the change in study
endpoint. The FDA describes phase II trials as follows: “Phase II includes
controlled clinical studies conducted to evaluate the effectiveness of the drug
for a particular indication or indications in patients with the disease or
condition under study and to determine the common or short term side effects and
risks associated with the drug. Phase II studies are typically well-controlled,
closely monitored, and conducted in a relatively small number of patients.”
Whereas the phase I studies were usually conducted with an open design, the
phase II studies are randomized. One or more groups of patients, depending on
the number of doses being studied, will receive the study drug while another
group, the control group, will receive a placebo with or without standard
therapy. These studies are usually double-blind; neither the patient nor the
investigator knows who is getting test drug, what dose of test drug is being
administered and who is getting placebo. This increases the complexity of the
operational aspects of drug packaging and allocation of treatments. The
investigational drug must be packaged in an identical manner to the placebo and
which must be given to the patients in accordance with a pre-determined
randomization code.
The overall aim of the phase II studies is to defi ne the type of
studies that will be conducted in phase III, including dose, duration of dosing,
frequency of dosing, patient population, and primary variable. When this
evaluation is poorly conducted, the phase III outcome becomes much more of a
guessing game than is necessary. Additional time spent in phase II to establish
the best design for phase III is likely to be returned by a shorter successful
phase III. Companies that truncate phase II so that they can start pivotal phase
III studies as early as possible do so at their own peril. The end result is
often a case of more haste, less speed. Once phase II is completed, the option
exists to meet with the FDA before starting phase III. The FDA regulations state
that the purpose of the end-of-phase II meeting is:
• To determine the safety of proceeding to phase III
• To evaluate the phase III plan and protocols
• To identify any additional information necessary to support a
marketing application for the uses under investigation.
As the third bullet point suggests, this meeting is not confined
to evaluating only the phase III program. The FDA defi nition of the
end-of-phase II meeting states that the focus should be “directed primarily at
establishing agreements between FDA and the sponsor of the overall plan for
phase III and the objectives and design of particular studies. The adequacy of
technical information to support phase III studies and/or a marketing
application may also be discussed.” Phase II is usually conducted in a few
hundred patients. Of the seven out of 10 drugs that complete phase I
successfully, only four will complete phase II and advance to phase III.
Phase III
The studies conducted in phase III are the NDA-enabling studies.
In the Federal Food, Drug and Cosmetic Act of 1938 (FFDCA), Drug Amendments of
1962, language regarding clinical study approval requirements read: “…adequate
and well controlled investigations…,” which was interpreted by the FDA as a
minimum of two such phase III studies. In the mid-1990s Carl Peck, M.D., the
ex-director of the FDA Center for Drug Evaluation Research, who had left the FDA
to start the Center for Drug Development Science, at Georgetown University in
Washington, DC, questioned the validity and utility of this interpretation. Over
the next few years this evolved into the concept of single clinical trial
submissions, where only one adequate and well-controlled phase III study may be
required in certain circumstances.
In 1997, President William Jefferson Clinton signed the Food and
Drug Administration Modernization Act. The language in Section 115a related to
the evidence of effectiveness required to approve a new drug. It was
substantially different from the FFDCA with Drug Amendment of 1962: “…data from
one adequate and well-controlled investigation and confirmatory evidence.” The
implication is clear; it is no longer mandatory that two adequate and
well-controlled studies (i.e., two phase III studies) are necessary for proof of
effectiveness required to approve a new drug. A debate still rages over what
constitutes “confirmatory evidence.”
One option for the basis of regulatory approval is a single phase
III clinical study plus causal confirmation.
Causal confirmation
has been described by Peck et al. as proof that “the
drug, through its pharmacological action, favorably alters the clinical
condition of those who are treated with it.” In other words, proof of
pathophysiologic and pharmacologic mechanisms is sufficient to be considered
confirmatory. Simplistically,
approval to market a drug could be based on one positive, adequate, and
well-controlled phase III study supported by a phase II study where the endpoint
was a surrogate endpoint marker. For example, if it is accepted that chest x-ray
fi ndings may be indicative of pulmonary function deterioration and the claim
for the drug will be preservation of pulmonary function, one adequate and
well-controlled phase III study with a pulmonary function endpoint supported by
one phase II chest x-ray positive finding adequately fulfills the requirements
of the act. There are a number of
additional questions that should be addressed in the phase III program program.
The primary requirement is to confirm the findings of safety and efficacy in the
phase II program. Inclusion and exclusion criteria to defi ne the study patient
population tend to be less rigorous in the phase III program, so the patients
more closely resemble the population who will be administered the drug when it
is marketed. For chronic treatments there is a requirement to dose for longer
periods than are normally considered in phase II. The ICH in its efficacy
guideline E1 has determined that the exposure to assess clinical safety for drug
intended for chronic administration in non-life-threatening conditions should be
at lease 500 patients for six months and 100 patients for one year.
Different drugs require different programs to develop sufficient
evidence to gain marketing approval. Special population clinical study
requirements are important (e.g., the elderly or children). Ultimately, the aim
of this gargantuan clinical development program is to establish sufficient
cause, safety, and efficacy to allow a new drug to be added to the physician’s
therapeutic armamentarium for the benefit of patients. The total patient
population required to complete phase III is highly variable. The ICH states
that a minimum of 1500 patients should be exposed to the drug before a marketing
application should be assessed, but that may be wholly inadequate in terms of
the number of patients who might be required in clinical trials to satisfy the
statistical considerations for proof of efficacy. Alternatively, in rare
conditions, dealt with in 21 CFR 360.20, Orphan Drug Regulations, significantly
fewer patients may be required to satisfy the phase III commitment. (Orphan drug
status in the United States can be sought for a drug intended for diseases or
conditions affecting less than 200,000 patients per year. In Europe the patient
population should not exceed 185,000 patients per year, and in Japan, 50,000
patients per year.) Approximately 70 to 90% of drugs that enter phase III
eventually make it to the market.
Phase IV
There is one additional phase of drug development which is
unrelated to providing documentation for the initial NDA. This is called phase
IV. These studies are run to generate additional data on the drug that are not
necessary for the approval process but might be used to modify the label text.
In phase IV the following information may be sought:
• Relative efficacy compared to different drugs used to treat the
same disease
• Cost-effectiveness of the drug
• Assessment of the improvement in the patient’s quality of life
• Safety assessment in an unselected patient population
• Opportunities for additional indications
The clinical development of drugs is usually defi ned as
sequential testing through phases I to III for approval and phase IV for
additional definition. The process, however, should not be thought of as
strictly sequential development. In ICH guideline E8, General Considerations for
Clinical Trials, an alternative nomenclature is proposed which more
appropriately reflects the development process. Whereas it is usually correct to
say that phase II does not start until an adequate phase I is conducted and
phase III will not begin until an appropriate phase II has been completed, just
because the drug is being evaluated in phase III does not mean that assessments
usually associated with phases I and II are not continuing to be developed.
E8 defines the development process as follows:
• Human pharmacology
(roughly equivalent to phase I)
• Assess tolerance.
• Defi ne pharmacokinetics and pharmacodynamics.
• Estimate activity.
• Therapeutic exploratory
(roughly equivalent to phase II)
• Explore use for targeted indication.
• Defi ne dose range for subsequent studies.
• Generate data to determine study designs.
• Therapeutic confi rmatory
(roughly equivalent to phase III)
• Confi rm efficacy.
• Demonstrate safety as well as can be defi ned within the
limited exposure of a
clinical program.
• Establish a dose–response relationship.
• Therapeutic use
(roughly equivalent to phase IV)
• Refi ne risk–benefit relationship.
• Identify less common adverse reactions.
• Refi ne dosing recommendations.
The concept of a logical serial development of a drug has much to
commend it, but frequently there are misunderstanding as to the actual stage of
development by misinterpreting the underlying basis of the four temporal phases.
Frequently, in the more sophisticated development programs there may be as much
or more human pharmacology assessment during phase III as there was during phase
I.
When a hypothesis has been developed, when a chemical synthesis
program has been developed, when an exploratory preclinical program has been
completed, and when the clinical study evaluation all contribute to the data
that generates an approved NDA, all the disciplines of research and development
come together to provide a tool to the physician to improve the life of his or
her patients.